




| Original Article | ||
Open Vet J. 2020; 10(4): 363-370 doi: 10.4314/ovj.v10i4.2 Open Veterinary Journal, (2020), Vol. 10(4): 363–370 Original Research http://dx.doi.org/10.4314/ovj.v10i4.2 Metabolic markers of myocardium insulin resistance in dogs with heart failureOleynikov Dmitrij Arkadievich1,2*1Almazov National Medical Research Center, Petersburg, Russia 2Veterinary Clinic “Belij Klyk”, Moscow, Russia *Corresponding Author: Oleynikov Dmitrij Arkadievich. Almazov National Medical Research Center, IEM, St. Petersburg, Russia. Email: wolfberg.guard [at] gmail.com Submitted: 26/08/2020 Accepted: 21/09/2020 Published: 20/10/2020 © 2020 Open Veterinary Journal
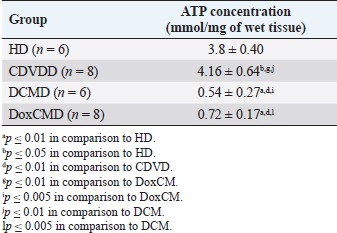
AbstractBackground: Heart failure syndrome is an aspect of primary or secondary heart disease and is associated with decompensation, formation, and activation of pathological interactions between regulation systems. This results in myocardial energy metabolism alteration. This study was carried out to defy some metabolic aspects of myocardial tissue insulin resistance (IRM) development in canine heart failure. Aim: To investigate the myocardial tissue concentration of adenosine triphosphate (ATP), glucose transporters 1 and 4, pyruvate dehydrogenase (PDH), hexokinase 2, insulin receptor (InsR), and adropin (ADR) protein and to screen metabolic changes and IRM in canine myocardium with heart failure. Methods: We studied 28 dogs of different sexes, ages, and breeds. Groups were formed according to primary pathology: apparently healthy dogs (HD, n=6); dogs with CDVD (CDVDD, n=8); dogs with DCM (DCMD, n=6); and dogs with doxorubicin chemotherapy and doxorubicin-induced cardiomyopathy (DoxCMD, n=8). Animals in the study were diagnosed for primary disease by standard methods and algorithms. Animals were euthanized due to incurable neurological disease, refractory heart failure, or by owners will. The material was obtained immediately after death, fixed in liquid nitrogen, and stored in −80°C refrigerator. Studied proteins concentrations were analyzed in a specialized research laboratory, using ELISA kits, provided by Cloud-Clone Corp. Results: ATP, GLUT1, and GLUT4 concentrations in myocardial tissue from the valvular disease group did not differ from the HD group. In CDVD, we found depression of PDH, hexokinase II (HX2), and ADR concentrations in comparison to HD. InsR was significantly lower in the CDVD and DoxCMD groups in comparison to the HD group, but in the DCM group, it was twofold higher than in the HD group. In the DCMD and DoxCMD groups, all parameters were lower than in the HD group. ATP, HX2, ADR, GLUT1, and GLUT4 were higher in the CDVD group, than in the DCM and DoxCM groups. PDH in the CDVD and DoxCM groups did not differ. PDH was depleted in the DCM to CDVD and DoxCM groups. InsR did not differ between the CDVD and DoxCM groups, but was upregulated in the DCM to CDVD and DoxCM groups. Conclusion: Development of myocardial tissue IRM is a part of the structural, functional and metabolic remodeling in dogs with heart failure of different etiology. At the late stages, we found significant changes in energy supply availability and production in the myocardium. Keywords: Canine, Cardiomyopathy, Heart failure, Insulin resistance, Myocardial metabolism. Introduction Heart failure syndrome is an aspect of primary or secondary heart disease and is associated with decompensation, formation, and activation of pathological interactions between components of neurohumoral regulation systems. This state is also characterized by myocardial energy metabolism suppression (Lopaschuk et al., 2010). For example, systolic dysfunction is associated with adrenal system activation, leading to heart rate acceleration. This catecholamine-induced action provokes increased oxygen demand in the myocardium, increased free fatty acids (FFA) consumption as an energy resource, adenosine diphosphate (ADP) accumulation, and negative inotropic effect. These conditions are well observed in chronic sympathetic hyperactivity in heart failure (Opie et al., 1979; Liu et al., 2001; Neubauer, 2007; Scwenk and Luiken, 2008; Fukushima and Lopaschuk, 2016). The main substrates for adenosine triphosphate (ATP) formation in the myocardium are carbohydrates and FFA (Lopaschuk et al., 2010). The energy resources include the following: triglycerides (TG), long-chained fatty acids, glucose, glycogen, lactate, pyruvate, ketone bodies (acetoacetate and beta-hydroxybutyrate) and in some cases amino acids (leucine, valine, and isoleucine). Listed substrates are degraded to intermediates, which then are included to Kreb’s cycle as an acetyl-coenzyme A (ACoA) or as a metabolic equivalent. During the utilization of these substrates a proton is generated, forming an electrical potential between mitochondrial membranes, and ADP to ATP is phosphorylated through the respiratory chain (Tian and Abel, 2001; Ingwall, 2009; Shibayama et al., 2015). This dispersion in substrates for uniform energy fuel is a base for several concepts: (1) myocardial metabolism is adaptive to the whole organism state and substrates are available and can be autonomically changed for more suitable energy supply; however, in heart failure this flexibility is limited; (2) myocardial metabolism is automatically regulated and all metabolic intermediates are mediators controlling metabolism pathways and their intensity (Randle’s cycle); (3) intermediates can be used for cell structure synthesis and, at the same time, degrading cell’s components can be utilized as energy resource; (4) metabolic alterations and intermediates accumulation can damage cells’ proteins and alter the ability of cardiomyocytes to contract; (5) myocardial metabolism is not “cell chemistry”, but it is a functional unit with its structure and mediators which controls cardiomyocytes adaptability (Taylor et al., 2001; Knupfer and Beckstein, 2013). The diversity of heart failure etiology in dogs has a wide specter. The most common diseases that cause heart failure in dogs are chronic myxomatous degenerative valvular disease (CMDV, CDVD, and MVD), dilated cardiomyopathy (DCM), arrhythmogenic right ventricular cardiomyopathy, primary or secondary pulmonary arterial hypertension, myocarditis, toxic alterations (doxorubicin), and inherited diseases. Most of them are characterized by cardiac chambers dilatation, systolic and diastolic dysfunction, and associated arrhythmias. In this study, we tried to defy some metabolic aspects and markers of myocardial tissue IRM development in most common heart diseases of dogs (CDVD, DCM, and doxorubicin-induced cardiomyopathy/DoxCMD) and compare them with clinically healthy animals to elucidate myocardial metabolic remodeling occurring in heart failure. Material and MethodsIn this work, we studied 28 dogs of different sexes, ages, and breeds. All dogs were patients of veterinary clinics in Saint Petersburg, Russia. Groups were formed according to primary pathology: apparently healthy dogs (HD, n=6); dogs with CDVD (CDVDD, n=8); dogs with DCM (DCMD, n=6); and dogs with doxorubicin chemotherapy (DoxCMD, n=8). This study aims to elucidate some metabolic aspects attributed to glycolysis and glucose oxidation of each disease. The object of the study is the myocardium. The material was obtained immediately after death. Samples for the metabolic study were taken from the apical part of the left ventricular free wall and fixed in liquid nitrogen at once and were stored in −80°C refrigerator. Animals in the study were diagnosed for primary disease by standard methods and algorithms (clinical findings, anamnesis, blood analyses, radiography, echocardiography, electrocardiography, and blood pressure measurement). Patients without signs of heart failure were euthanized due to the low quality of life (most of them had primary neurological disease). Dogs from the other groups were euthanized due to refractory heart failure, unsuccessful neoplasia treatment, or by owners’ will due to low quality of life. Obtaining samples was agreed upon by the owners. For biochemistry analysis, we used ELISA kits, provided by Cloud-Clone Corp. (Katy, TX), which included the following: total ATP, pyruvate dehydrogenase (PDH), hexokinase II (HX2), Adropin (ADR), GLUT1 and GLUT4, and insulin receptor (InsR). Statistical analysis was carried out on STATISTICA 7.0 software, using Fisher’s exact test and Mann–Whitney criteria. Ethical approvalThe scheme of this study was ethically approved on clinical conference and by Research Standard department of Almazov National Medical Research Center. ResultsThe group of dogs without cardiac disease (HD) included animals with absent signs of heart dysfunction observed by echocardiography and electrocardiography methods. The reasons for euthanasia in this group were neurological diseases (epilepsy, local traumas of the cervical and thoracic spine, and neoplasm of brain and spinal cord), where the treatment was unsuccessful or was associated with low quality of life. Euthanasia was carried out with owners’ will. Results obtained from biochemical studies were used as criteria of apparently physiological condition. In parallel, the data were compared with available information from studies of other species, and our previous studies on myocardial metabolomics in dogs with heart failure. Observed ATP concentration in the HD group showed a sufficient amount of macroergic phosphates for myocardium contractility energy supply (Table 1). These data converge with metabolomic studies from rats and our previous results. Table 1. ATP concentration in the myocardium.
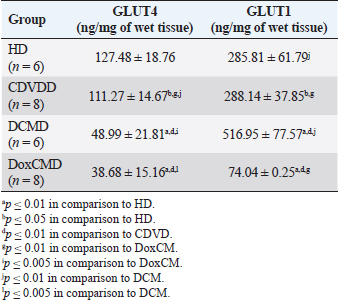
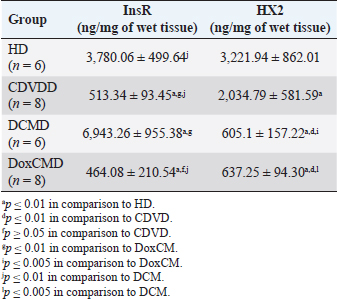
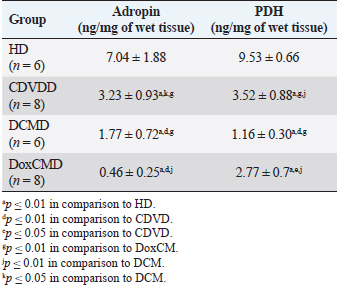
Unfortunately, the physiological concentration of Adropin, PDH, HX2, GLUT1, and InsR for dogs is not described in the available literature. Due to this fact, we accepted that information obtained from the HD group as it is mostly unaltered and characterizes physiological condition (Tables 2–4). ATP concentration in myocardial tissue from the CDVD group did not differ from this parameter in the HD group (p ≥ 0.05). Additionally, GLUT4 and GLUT1 protein concentrations were similar to these parameters in HD (p ≥ 0.05 for both). In other CDVD group measurements, we discovered the statistically strong depression of PDH, HX2, ADR, and InsR (p ≤ 0.01, for each) in myocardial tissue in comparison to the HD group. Table 2. GLUT1 and GLUT4 concentrations in the myocardium.
Table 3. InsR protein and Hexokinase2 concentrations in the myocardium.
All parameters observed in the DCMD and the DoxCMD groups were statistically lower than that in the HD group (p ≤ 0.01, except GLUT1 for DCMD). Nonetheless, there were differences in heart failure groups. ATP concentration was significantly higher in the CDVD group than in the DCMD and DoxCMD groups (p ≤ 0.01, for both), but this parameter did not differ between DCMD and DoxCMD group (p ≥ 0.05). As for PDH, we observed the non-statistically significant difference between the concentration of the protein in the CDVD and DoxCMD groups (p ≤ 0.05) and, at the same time, PDH concentration was severely depleted in DCM group to CDVD (p ≤ 0.01) and DoxCMD (p ≤ 0.01) groups. HX2 protein concentration in myocardial tissue was statistically lower in both DCM and DoxCMD groups than in CDVD group (p ≤ 0.01), but did not significantly differ between each other (p ≥ 0.05). Adropin was depleted in the DCMD group in comparison to CDVD (p ≤ 0.05), but in the DoxCMD group, there was a great lack of the protein comparing both with DCMD and CDVD (p ≤ 0.01) groups. As for glucose transport system, we found that GLUT4 concentration was significantly depleted in both DCMD and DoxCMD groups compared to CDVD (p ≤ 0.01); at the same time, we observed lower concentration of this protein in DoxCMD group in comparison to DCMD group, but this fact was not statistically proven (p ≥ 0.05). GLUT1 proteins were severely depleted in both DoxCMD to CDVD (p ≤ 0.01) group; also, GLUT1 was significantly lower in DoxCMD to DCMD (p ≤ 0.01). In the DCMD group, GLUT1 was significantly increased to CDVD and HD (p ≤ 0.01). InsR protein differed non-significantly between CDVD and DoxCMD groups (p ≥ 0.05). This parameter was several times higher in the DCMD group in comparison to the CDVD, HD, and DoxCMD groups (p ≤ 0.01). Table 4. Adropin and PDH concentrations in the myocardium.
DiscussionIn this study, we did not find significant differences between HD and CDVDD groups in ATP concentration. This could be explained by the fact, that heart failure associated with degenerative valvular disease does not have enough influence to alter the energy supply in the myocardium. Thus, we can suppose metabolic dysregulation does not play the main role in the development of systolic and diastolic dysfunction, it mostly depends on volume overload and decompensation in the Frank–Starling mechanism (Mercadier et al., 1987; Komamura et al., 1994). From heart failure studies we can see adaptive changes in metabolic and protein profiles, which lead to increased production of heavy chains of myosin and BB-isomer of creatine kinase (CK). These changes are a part of the shift to fetal metabolic profile, which helps to sustain effective work in conditions of decreased oxygen and energy supply. Moreover, the part of this shift is imbalanced between the expression of GLUT1 and GLUT4. This could be explained by two models: (1) decreased insulin sensitivity leads to suppression of GLUT4 trafficking to a membrane (although the protein concentration is still stable and we see this in cases of CDVD); (2) absolute depletion of GLUT4 proteins and a compensatory increase in GLUT1 (which is still a part of tissue IRM on late stages) (Schulz et al., 1997; Friehs et al., 1999; Tokarska-Schlattner et al., 2005; Kolwitcz and Tian, 2011; Taegetmeyer et al., 2016). In our study, we did not recognize changes in GLUT4 and GLUT1 expressions in comparison to HD group. In the previously mentioned concept of IRM, there is a fact of unchanged expression of proteins or microRNA of GLUT4, despite alterations in exocytosis of transporting molecules (Friehs et al., 1999; Ventura-Clapier, 2004; Fazakerley et al., 2009). In our study, we did not elucidate the factor of GLUT4 trafficking, but this concept could be a marker of a silent or hidden IRM in terminal stages of CDVD-dependent heart failure. In the CDVD group, we found a significant decrease in InsR expression in comparison to HD. Therefore, we can suppose that this can influence on IRM development due to decreased intensity of GLUT4 exocytosis (without absolute protein concentration changes) and suppression of glucose utilization. This is also connected with the suppression of PDH and HX2 expression in myocardial tissue. The depletion of intermediates of glucose metabolism firstly leads to suppression of HX2 synthesis, then to decreased availability of pyruvate suppress PDH and such depletion activates beta-oxidation of FFA. This kind of IRM development should be monitored as a complicated mechanism of interactions: increased demand in energy substrates, stromal myocardial remodeling, changes in vascular architecture and oxygen delivery, adrenal system overactivation, and possibly, parasympathetic suppression. Human medicine defines that IRM is developed during the stage of New York Heart Association classification and adrenaline/noradrenaline effects are significant (Stanley et al., 2005; Ashrafian et al., 2007). During heart failure development, beta-oxidation of FFA as a main resource of energy accentuates, leading to migrating GLUT4 availability and suppressing of genes associated with InsR, PDH and GLUT4 resynthesis (Schulz et al., 1997; Ashrafian et al., 2007). Moreover, during the first stages, exocytosis of GLUT4 is increased, but then endocytosis becomes altered (this is a side effect of catecholamines) (Yang and Holman, 2005; He and Liu, 2007). This pathogenetic mapping is supported by ADR changes. Almost a twofold-decreased ADR concentration elucidates IRM. Therefore, decreased concentrations of InsR, PDH, and HX2 increased FFA consumption, GLUT4 translocation inhibition, leading to ADR secretion suppression by feedback principle (Aydin et al., 2015). To summarize, in the CDVD group the main parameters of myocardial metabolism were stable, according to our previous study (Oleynikov, 2017). However, in detail, we found markers of developing IRM, which decreases liability of myocardial energy supply and worsen the adaptation to heart failure. Next, we observed several characteristics of DoxCMD myocardial metabolism. Energy production in DoxCMD is severally suppressed which is sustained by extremely low concentrations of ATP. This alteration is associated with multistage failure in metabolism. First of all, we analyze aspects of the glycolytic system. Previously, we described changes in myocardial lactate and pyruvate concentrations, LDH and general CK activity (Oleynikov, 2017). These results showed depressed glucose utilization and a decrease in intermediates production. This was the base of our opinion of PDH block due to Randle’s cycle activation (Randle et al., 1963; Scwenk and Luiken, 2008; Lopaschuk et al., 2010). This is sustained by the fact that tissue lactate level characterizes glycolysis intensity and metabolites exocytosis. However, decreased glucose traffic to the cell leads to suppression of intermediates production (especially glucose-6-phosphate) and the suppressing of HX2 activity. Hexokinase is one of the threshold enzymes for glucose metabolism, because HX activity stays unchanged, which is a reason of glycolysis limitation in energy supply despite its increased demand (due to energy starvation) and microRNA expression (Ritchie and Delbridge, 2005; Lionetti et al., 2007; Scwenk and Luiken, 2008; Ardehali et al., 2012; Taegetmeyer et al., 2016). Decreased glucose utilization leads to depletion in pyruvate for oxidation. This condition is associated with Kreb’s cycle alteration, due to metabolites “loss” (fumarate, succinate, malate, oxoglutarate) and acceleration of this process during heart failure, insufficiency of pyruvate decreases the ability to sustain the number of lost metabolites (Scwenk and Luiken, 2008). Pyruvate insufficiency also leads to decreased lactate production and changes in the redox potential of cytoplasm. Alteration of NAD/NADH ratio (associated with suppression of NAD-dependent enzymes) elucidates a decrease of macroergic phosphates production. In conditions of suppressed ATP production, the activity of NAD utilization and restoration decreases and negatively affects enzyme activity. Also increased NAD could be a consequence of mitochondria damage due to oxidative stress (Jang et al., 2012). The study showed that doxorubicin can damage mitochondrial complex 1 and provoke hydroxyl radicals overproduction (Daviest and Doroshow, 1986; Weinstein et al., 2000). Therefore, we can suppose two reasons for decreased LDH activity: depleted glycolysis and Dox effect on NAD-dependent dehydrogenase systems (inability to restore pyruvate from available lactate) (Garcia et al., 1994; Johannsson et al., 1997; Van der Vusse et al., 2000; Outomuro et al., 2007). In heart failure, there is an adrenal system overactivation that leads to increased catecholamines circulation and, as a consequence, lipomobilisation and increased FFA. Overutilization of FFA in myocardial metabolism leads to PDH and glycolysis block. In the case of DoxCM, this is worsened by suppression of CPT- and NAD-dependent systems. This associated with slowed utilization of cytosolic FFA and conversion of them to the TG. They are stored as lipid droplets in the cytosol. While the demand for energy substrates stays high and glycolysis is blocked, these droplets are included in “energy–waste” cycles of degradation and stabilization, with side production of ceramides, ROS, and activation of uncoupling protein 3 (energy production is dismissed by heat production) (Opie et al., 1979; Sack et al., 1996; Taylor et al., 2001; Scwenk and Luiken, 2008; Jastroch et al., 2010). In parallel, Dox alters CK activity, blocking its connection to the outer mitochondrial membrane and transport of ATP to creatine (Wallimann et al., 1992; Tokarska-Schlattner et al., 2005; Taegetmeyer et al., 2016). We also found the depletion of GLUT1 and GLUT4 proteins, which is the reason for decreased activity PDH, HX2, and development of tissue IRM (Randle et al., 1963; Schulz et al., 1997). Decreased PDH and HX2 show not only a metabolic remodeling but also a significant decrease in glucose intracellular transport, leading to feedback decrease in the synthesis of HX2, pyruvate production, cytosol acidosis, and PDH deactivation (Razeghi and Young, 2001). In the study of Mouline et al. (2015), DoxCMD was associated with decreased activity of AMPK, ACoA carboxylase, suppression of GLUT4, and HX2 (correlated with AMPK depression). In parallel, PDK4 activity was increased, which is associated with PDH activity block and decrease of glucose utilization (Korvald, 2000; Ashour et al., 2012; Mouline et al., 2015). In experimental work with rats, the DoxCM-associated IRM was admitted, and was associated with decreased microRNA and proteins of GLUT4 and AMPK. This could be explained by Dox-assisted inhibition of InsR and GLUT interactions (de Lima et al., 2016). In the DoxCMD group, InsR was depleted too, which leads to changes in the synthesis of Insulin–InsR–GLUT4 axis proteins. To sum up, FFA oxidation overactivation and Dox effect (PDH, HX2, and mitochondria) cause changes in GLUT trafficking providing IRM, resulting in InsR suppression and finishing in stable IRM vicious circle. Adropin is a new marker of tissue glucose metabolism. It is produced in the heart and could be a specific marker of tissue IRM development. In some studies, ADR is associated with heart failure stage. In our study, we tried to determine connection between development of heart failure and changes in ADR expression. ADR was significantly depleted in DoxCMD and could be estimated as a consequence of metabolic decompensation. This data is consistent with the tendency admitted to heart failure but differs from available sources. One of the studies, elucidating changes in copeptin, irisin, and adropin under Dox effects, showed increased ADR concentrations in myocardium and plasma on the 14th day of the experiment (Aydin and Kuloglu, 2013). In our study, we defined a tremendous decrease of ADR in the myocardium. It could be explained by some facts: doses (cumulative dose was higher in mg/kg), chronic mechanism of heart failure onset. A described experiment on rats is an example of fast developed heart metabolic changes, while our work is a presentation of chronic heart failure due to the Dox-prolonged exposition. In the DCMD group, we similarly admitted significant changes in energy supply. In our previous research, we found a slight decrease in lactate concentration and LDH activity (Nikolaidis et al., 2004). This could be explained by relatively saved glucose metabolism, due to the absence of additive Dox damage. Also, we admit that, despite the decrease of GLUT4 proteins in comparison to HD and CDVDD, GLUT4 was higher than in DoxCMD and increased InsR. This data can be explained as a marker of IRM development because energy starvation provides an increased expression of InsR, but active FFA oxidation suppresses GLUT4 production and alteration of the Insulin–InsR–GLUT4 axis. The glucose flux is modulated by increased GLUT1 expression. GLUT1 is a weak insulin-dependent transporter, but insulin promotes GLUT1 synthesis and sustains intercellular glucose flux. These modifications cannot transport enough resources for adequate glucose metabolism. In experimental works on dogs with DCM, GLUT4 expressions did not change, but in studies with chronic infarct, this protein was severely decreased (Nikolaidis et al., 2004). In Nikolaidis’s study, tissue IRM was also admitted; markers of this state were associated with decreased FFA and glucose consumption and increased oxygen demand (desynchronization of glycolysis and oxidative phosphorylation). In this study, GLUT4 protein expression in tissues decreased; this fact correlates with increased circulated FFA, their utilization, and suppression of GLUT4 resynthesis being the feedback principle of Randle’s cycle. The data could be associated with high tonus of the sympathetic nervous system, associated catecholamine-mediated lipolysis, and AMPK activation (Nikolaidis et al., 2004). The observed differences in these experiments could be explained by the fact, that Nikolaidis’s study was carried out during the early stages of heart failure (35–42 from DCM modulation) when the metabolic alteration was not developed. In Murray’s study, metabolic alterations seemed more prominent and this could be explained by the length of the period (70 days) (Murray et al., 2006). In our study, dogs with the diagnosis of DCM were treated for about 200 days. Prolonged active FFA utilization provides PDH and HX2 suppression. As it was previously mentioned, HX2 is a limiting threshold for the Insulin–InsR–GLUT4 axis, this state decreases the number of glycolysis metabolites (pyruvate). This condition leads to further activation of FFA utilization, PDH block (due to PDK4 activation), decrease in pyruvate utilization, and its transformation to lactate (this metabolite was mostly unchanged in our previous study). Adropin was decreased in the DCMD group in comparison to HD and CDVD groups, but was higher than that in DoxCMD group. This fact could be explained by stimulating the role of ADR for glucose metabolism, as an alternative source of energy, but a long period of disease and development of IRM is the reason for the suppressed expression of ADR. ConclusionIn conclusion, changes in myocardial metabolism associated with heart failure in dogs were described. Development of myocardial tissue IRM is a part of structural, functional, and metabolic remodeling in dogs with heart failure of different etiology. At the late stages, significant changes in energy supply availability and production in the myocardium were found. Further studies are needed to identify plasma markers for metabolic alteration, possible role, and treatment. ReferencesArdehali, H., Sabbah, H.N., Burke, M.A., Sarma, S., Liu, P.P., Cleland, J.G. 2012. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. Eur. J. Heart Fail. 14, 120–129. Ashour, A.E., Sayed-Ahmed, M.M., Abd-Allah, A.R., Korashy, H.M., Maayah, Z.H., Alkhalidi, H., Mubarak, M. and Alhaider, A. 2012. Metformin rescues the myocardium from doxorubicin induced energy starvation and mitochondrial damage in rats. Oxid. Med. Cell. Longev. 2012, 434195. Ashrafian, H., Frennaux, M.P. and Opie, L.H. 2007. Metabolic mechanisms in heart failure. Circ. 116, 434–448. Aydin, S. and Kuloglu, T. 2013. Expression of adropin in rat brain, cerebellum, kidneys, heart, liver and pancreas in streptozotocin-induced diabetes. Mol. Cell. Biochem. 380, 73–81. Aydin, S., Eren, M. and Kuloglu, T. 2015. Alteration of serum and cardiac tissue adropin, copeptin, irisin and TRPM2 expressions in Dox treated male rats. Biotech Histochem. 3, 197–205. Daviest, K. and Doroshow, J. 1986. Redox cycling of anthracyclines by cardiac mitochondria. J. Biol. Chem. 261, 3060–3067. de Lima, J., Yamashita, A. and Pimentel, G. 2016. Doxorubicin caused severe hyperglycaemia and insulin resistance, mediated by inhibition in AMPK signaling in skeletal muscle. J. Cachexia. Sarcopenia. Muscle. 7, 615–625. Fazakerley, D.J., Lawrence, S.P. and Lizunov, V.A. 2009. A common trafficking route for GLUT4 in cardiomyocytes in response to insulin, contraction and energy-status signaling. J. Cell Sci. 122, 727–734. Friehs, I., Moran, A.M., Stamm, C., Colan, S., Takeuchi, K., Cao-Danh, H., Rader, C., McGowan, F. and del Nido, P. 1999. Impaired glucose transporter activity in pressure overload hypertrophy is an early indicator of progression failure. Circulation. 100, 187–193. Fukushima, A. and Lopaschuk, G. 2016. Acetylation control of cardiac fatty acid b-oxidation and energy metabolism in obesity, diabetes and heart failure. Biochim. Biophys. Acta. 1862, 2311–3220. Garcia, C.K., Goldstein, J.L., Pathak, R.K., Anderson, R.G. and Brown, M.S. 1994. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: implications for the Cori cycle. Cell. 76, 865–873. He, A. and Liu, X. 2007. How many signals impinge on GLUT4 activation by insulin? Cell. Signal. 19, 1–7. Ingwall, J.S. 2009. Energy metabolism in heart failure and remodelling. Cardiovasc. Res. 81, 412–419. Jang, S., Tae Kang, H. and Seong Hwang, E. 2012. Nicotinamide-induced mitophagy event mediated by hgh NAD/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 23, 19304–19314. Jastroch, M., Divakaruni, A.S., Mookerjee, S., Treberg, J.R. and Brand, M.D. 2010. Mitochondrial proton and electron leaks. Essays Biochem. 47, 53–67. Johannsson, E., Nagelhus, E.A., McCullagh, K.J., Sejersted, O.M., Blackstad, T.W., Bonen, A. and Ottersen, O.P. 1997. Cellular and subcellular expression of the monocarboxylate transporter MCT1 in rat heart. A high-resolution immunogold analysis. Circ. Res. 80, 400–407. Knupfer, C. and Beckstein, C. 2013. Structure, function and behavior of computational models in system biology. BMC Syst. Biol. 7, 43. Kolwitcz, S.C. and Tian, R. 2011. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 90, 194–201. Komamura, K., Shannon, R.P., Ihara, T., Shen, Y., Mirsky, I., Bishop, S.F. and Vatner, S.F. 1994. Exhaustion of Frank-Starling mechanism in conscious dogs with heart failure. Am. J. Physiol. Heart Circ. Physiol. 265, I119–1131. Korvald, C. 2000. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am. J. Physiol. Heart Circ. Physiol. 278, 1345–1351. Lionetti, V., Guiducci, L., Aquaro, G.D. 2007. Mismatch between uniform increase in cardiac glucose uptake and regional contractile dysfunction in pacing-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 293, 2747–2756. Liu, J., Wang, C., Murakami, Y. 2001. Mitochondrial ATPase and high-energy phosphates in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 281, 1319–1326. Lopaschuk, G.D., Ussher, J.R., Folmes, C.D., Jaswal, J.S. and Stanley, W.C. 2010. Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90, 207–258. Mercadier, J.J., De la Bastie, D., MBnasche, P., N’Guyen Van Cao, A., Bouveret, P., Lorente, P., Piwnica, A., Slama, R. and Schwartz, K. 1987. Alpha-myosin heavy chain isoform and atrial size in patients with various types of mitral valve dysfunction: a quantitative study. J. Am. Coll. Cardiol. 9, 1024–1030. Mouline, M., Piquereau, J. and Mateo, P. 2015. Sexual dimorphism of doxorubicin-mediated cardiotoxicity: potential role of energy metabolism remodeling. Circ. Heart Fail. 8(1), 98–108. Murray, A., Lygate, C. and Cole, M. 2006. Insulin resistance, abnormal energy metabolism and increased ischemic damage in the chronically infarcted rat heart. Cardiovasc. Res. 71, 149–157. Neubauer, S. 2007. The failing heart – an engine out of fuel. N. Engl. J. Med. 356, 1140–1151. Nikolaidis, L.A., Sturzu, A., Stolarski, C., Elahi, D., Shen, Y.T. and Shannon, R.P. 2004. The development of myocardial insulin resistance in conscious dogs with advanced dilated cardiomyopathy. Cardiovasc. Res. 61, 297–306. Oleynikov, D.A. 2017. Metabolic features of heart failure with different etiology. Am. J. Anim. Vet. Sci. 12, 32–44. Opie, L.H., Thandroyen, F.T., Muller, C. and Bricknell, O.L. 1979. Adrenaline-induced “oxygen-wastage” and enzyme release from working rat heart. Effects of calcium antagonism, beta-blockade, nicotinic acid and coronary artery ligation. J. Mol. Cell. Cardiol. 11, 1073–1094. Outomuro, D., Grana, D.R., Azzato, F. and Milei, J. 2007. Adriamycin-induced myocardial toxicity: new solutions for an old problem? Int. J. Cardiol. 117, 6–15. Randle, P.J., Garland, P.B., Hales, C.N. and Newsholme, E.A. 1963. The glucose fatty-acid cycle: its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1, 785–789. Razeghi, P. and Young, M. 2001. Metabolic gene expression in fetal and failing human heart. Circ. 11, 2923–2931. Ritchie, R. and Delbridge, L. 2005. Cardiac hypertrophy, substrate utilization and metabolic remodeling: cause or effect? Proc. Aust. Physiol. Soc. 36, 35–43. Sack, M.N., Rader, T.A., Park, S., Bastin, J., McCune, S.A. and Kelly, D.P. 1996. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circ. 94, 2837–2842. Schulz, R., Dodge, K.L., Lopaschuk, G.D. and Clanachan, A.S. 1997. Peroxynitrite impairs cardiac contractile function by decreasing cardiac efficiency. Am. J. Physiol. 272, 1212–1219. Scwenk, R.W. and Luiken, J.J.F.P. 2008. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc. Res. 79, 249–258. Shibayama, J., Yuzyuk, T.N., Cox, J., Makaju, A., Miller, M., Lichter, J., Hui, L., Leavy, J., Franklin, S., Zaitsev, A.V. 2015. Metabolic remodeling in moderate synchronous versus dyssynchronous pacing-induced heart failure; integrated metabolomics and proteomics study. PLoS One. 10, 32. Stanley, W.C., Recchia, F.A. and Lopaschuk, G.D. 2005. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85, 1093–1129. Taegetmeyer, H., Young, M. and Lopaschuk, G. 2016. Assessing cardiac metabolism. A scientific statement from the American Heart Association. Circ. Res. 118, 1659–1701. Taylor, M., Wallhaus, T.R., Degrado, T., Russell, D., Stanko, P., Nickles, R. and Stone, C. 2001. An evaluation of myocardial fatty acid and glucose uptake using PET with [18F] Fluoro-6-thia-heptadecanoic acid and [18F] FDG in patients with congestive heart failure. J. Nucl. Med. 42, 55–62. Tian, R. and Abel, E. 2001. Responses of GLUT4-deficient hearts to ischemia underscore the importance of glycolysis. Circ. 103, 2961–2966. Tokarska-Schlattner, M., Zaugg, M., da Silva, R., Lucchinetti, E. and Marcus, C., Wallimann, S.T. and Schlattner, U. 2005. Acute toxicity of doxorubicin on isolated perfused heart: response of kinases regulating energy supply. Am. J. Physiol. Heart Circ. Physiol. 289, 37–47. Van der Vusse, G.J., van Bilsen, M. and Glatz, J.F.C. 2000. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc. Res. 45, 279–293. Ventura-Clapier, R.F. 2004. Energy metabolism in heart failure. J. Physiol. 555, 1–13. Wallimann, T., Wyss, M., Brdiczka, D., Nicolay, K. and Eppenberger, H.M. 1992. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ’phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 281, 21–40. Weinstein, D.M., Mihm, J. and Bauer, J.A. 2000. Cardiac peroxynitrite formation and left ventricular dysfunction following doxorubicin treatment in mice. J. Exp. Pharmacol. 294, 396–401. Yang, J. and Holman, G. 2005. Insulin and contraction stimulate exocytosis, but increased AMPK activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J. Biol. Chem. 280, 4070–4078. | ||
| How to Cite this Article |
| Pubmed Style Oleynikov D, . Metabolic markers of myocardium insulin resistance in dogs with heart failure. Open Vet J. 2020; 10(4): 363-370. doi:10.4314/ovj.v10i4.2 Web Style Oleynikov D, . Metabolic markers of myocardium insulin resistance in dogs with heart failure. https://www.openveterinaryjournal.com/?mno=129120 [Access: April 26, 2024]. doi:10.4314/ovj.v10i4.2 AMA (American Medical Association) Style Oleynikov D, . Metabolic markers of myocardium insulin resistance in dogs with heart failure. Open Vet J. 2020; 10(4): 363-370. doi:10.4314/ovj.v10i4.2 Vancouver/ICMJE Style Oleynikov D, . Metabolic markers of myocardium insulin resistance in dogs with heart failure. Open Vet J. (2020), [cited April 26, 2024]; 10(4): 363-370. doi:10.4314/ovj.v10i4.2 Harvard Style Oleynikov, D. & (2020) Metabolic markers of myocardium insulin resistance in dogs with heart failure. Open Vet J, 10 (4), 363-370. doi:10.4314/ovj.v10i4.2 Turabian Style Oleynikov, Dmitrij, and . 2020. Metabolic markers of myocardium insulin resistance in dogs with heart failure. Open Veterinary Journal, 10 (4), 363-370. doi:10.4314/ovj.v10i4.2 Chicago Style Oleynikov, Dmitrij, and . "Metabolic markers of myocardium insulin resistance in dogs with heart failure." Open Veterinary Journal 10 (2020), 363-370. doi:10.4314/ovj.v10i4.2 MLA (The Modern Language Association) Style Oleynikov, Dmitrij, and . "Metabolic markers of myocardium insulin resistance in dogs with heart failure." Open Veterinary Journal 10.4 (2020), 363-370. Print. doi:10.4314/ovj.v10i4.2 APA (American Psychological Association) Style Oleynikov, D. & (2020) Metabolic markers of myocardium insulin resistance in dogs with heart failure. Open Veterinary Journal, 10 (4), 363-370. doi:10.4314/ovj.v10i4.2 |

