




| Research Article | ||
Open Vet. J.. 2024; 14(5): 1224-1242 Open Veterinary Journal, (2024), Vol. 14(5): 1224–1242 Research Article Immunoinformatics approach for designing a multiepitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike proteinElla Mae Joy S. Sira1, Edward C. Banico1, Nyzar Mabeth O. Odchimar1, Lauren Emily Fajardo1, Ferdinand F. Fremista Jr.2, Hanna Angelika B. Refuerzo2, Ana Patrisha A. Dictado2 and Fredmoore L. Orosco1,2,3*1Virology and Vaccine Research and Development Program, Industrial Technology Development Institute, Department of Science and Technology, Taguig City, Philippines 2Department of Biology, College of Arts and Sciences, University of the Philippines Manila, Philippines 3S&T Fellows Program, Department of Science and Technology, Taguig City, Philippines *Corresponding Author: Fredmoore L. Orosco. Virology and Vaccine Research and Development Program, Industrial Technology Development Institute, Department of Science and Technology, Taguig City, Philippines. Email: florosco [at] up.edu.ph Submitted: 20/03/2024 Accepted: 26/04/2024 Published: 31/05/2024 © 2024 Open Veterinary Journal
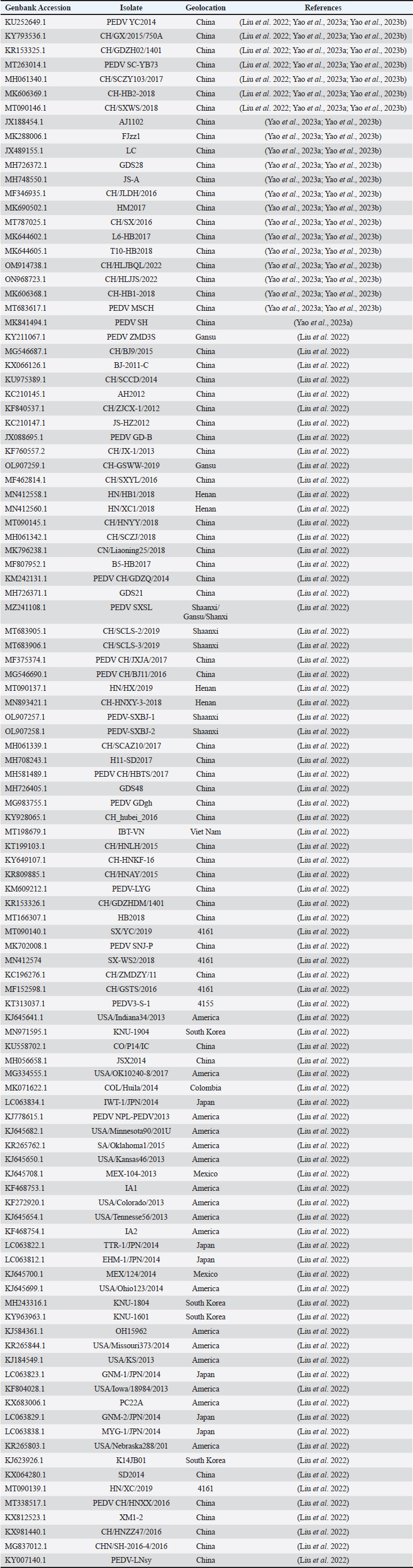
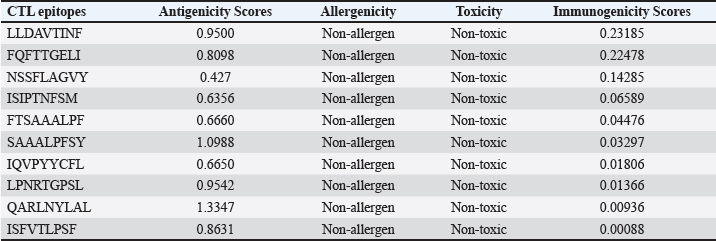
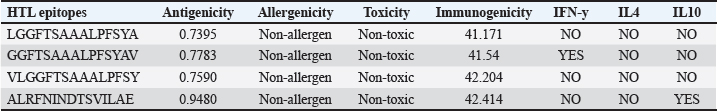
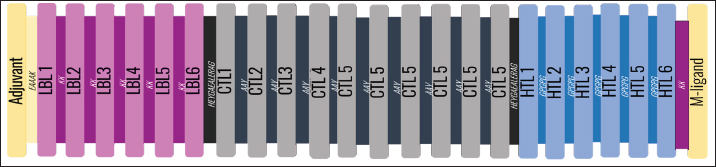
AbstractBackground: Porcine epidemic diarrhea (PED), caused by the porcine epidemic diarrhea virus (PEDV), is associated with high mortality and morbidity rates, especially in neonatal pigs. This has resulted in significant economic losses for the pig industry. PEDV genotype II-based vaccines were found to confer better immunity against both heterologous and homologous challenges; specifically, spike (S) proteins, which are known to play a significant role during infection, are ideal for vaccine development. Aim: This study aims to design a multi-epitope subunit vaccine targeting the S protein of the PEDV GIIa strain using an immunoinformatics approach. Methods: Various bioinformatics tools were used to predict HTL, CTL, and B-cell epitopes. The epitopes were connected using appropriate linkers and conjugated with the CTB adjuvant and M-ligand. The final multiepitope vaccine construct (fMEVc) was then docked to toll-like receptor 4 (TLR4). The stability of the fMEVc—TLR4 complex was then simulated using GROMACS. C-immsim was then used to predict the in vitro immune response of the fMEVc. Results: Six epitopes were predicted to induce antibody production, ten epitopes were predicted to induce CTL responses, and four epitopes were predicted to induce HTL responses. The assembled epitopes conjugated with the CTB adjuvant and M-ligand, fMEVc, is antigenic, non-allergenic, stable, and soluble. The construct showed a favorable binding affinity for TLR4, and the protein complex was shown to be stable through molecular dynamics simulations. A robust immune response was induced after immunization, as demonstrated through immune stimulation. Conclusion: In conclusion, the multi-epitope subunit vaccine construct for PEDV designed in this study exhibits promising antigenicity, stability, and immunogenicity, eliciting robust immune responses and suggesting its potential as a candidate for further vaccine development. Keywords: Immunoinformatics, Multiepitope subunit vaccine, PEDV, Reverse vaccinology. IntroductionPorcine epidemic diarrhea (PED) is an intestinal disease in pigs characterized by watery diarrhea, severe dehydration, vomiting, emaciation, and decreased reproductive performance. PED is caused by the porcine epidemic diarrhea virus (PEDV) belonging to the genus Alphacoronavirus within the family Coronaviridae, order Nidovirales. It affects pigs of all ages, with mortality, severity, and duration of the disease diminishing as age increases. Severe cases are associated with neonatal pigs, reaching up to 100% mortality and morbidity rate of 80%–100% causing serious economic losses to the pig industry worldwide (Salamat et al., 2021; Zhuang et al., 2022; Yu et al., 2023), necessitating the development of an effective protection strategy. Furthermore, the emergence of novel isolates and recombinant strains in Asia has led to recurrent outbreaks (Lin et al., 2022; Orosco, 2024a). PEDV was first detected in 1975 and has caused isolated and sporadic outbreaks in the Philippines. Outbreaks were recorded in 2005 in Cavite Laguna, Negros Occidental, Pampanga, Pangasinan, Quezon, and Tarlac. In 2006 and 2007, the Philippines had the highest mortality among China, Korea, and Vietnam, with an estimated 60,000 and 2,179 neonatal pig deaths, respectively (Salamat et al., 2021). Other recorded PED outbreaks in the Philippines include the 2010 outbreak in Batangas, which resulted in 11,414 swine deaths, and the 2016 outbreak in Agusan, Bacolod, Batangas, Bulacan, Cavite, General Santos, Laguna, Pampanga, Pangasinan, Rizal, and Tarlac, which killed most infected piglets, resulting in severe economic damage (Morales et al., 2007; Paraguison-Alili and Domingo, 2016). PEDV is an enveloped, positive-sense RNA virus that replicates in a porcine small intestine that has a genome approximately 28 kb in size. Its genome is composed of 5’ and 3’ untranslated regions; non-structural proteins such as open reading frame 1 ab (ORF1ab) polyprotein; structural proteins such as spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins; and an ion channel protein ORF3 (Salamat et al., 2021). Among these proteins, the spike (S) protein plays a significant role in cell infection and the immune response of the host. Furthermore, the presence of mutations in the S protein determines the PEDV genotype: genotype I (GI) or genotype 2 (GII). GII strains have mutations in their S protein, which makes them more virulent and epidemic. Specifically, highly virulent strains, such as PEDV strain CH/HLJJS/2022 from China, belong to the GIIa subtype (Yao et al. 2023a; Yao et al., 2023b). Sequence analysis of PEDV isolates in the Philippines revealed that both GI and GII strains are present in the country (Salamat et al., 2021). Vaccines against PEDV have been developed, including live vaccines such as attenuated and modified live vaccines (MLVs), and inactivated vaccines. However, these commercial vaccines only provide low-to-moderate defense against novel variants because of their genetic differences (Orosco, 2024a). Additionally, the continuous use of MLVs against PEDV could cause recombination between the vaccine and wild-type isolate to increase the survivability of the virus (Feng et al., 2023). Unlike the aforementioned vaccines, epitope-based vaccines can utilize short antigenic peptide fragments of the virus that can elicit an immune response through the production of specific antibodies that are more appropriate for viruses that have diverse variants. In terms of vaccine development, inactivated vaccine candidates based on the GIIa strain showed greater efficacy against GIIa and GIIb strains than candidates based on the GIIb strain (Liu et al., 2019). Moreover, GIIa-based vaccine candidates can provide better immunity against both heterologous and homologous PEDV challenges than GIIb candidates (Yu et al., 2023). Thus, utilizing GIIa strains could be useful in developing more effective vaccines. Finally, since PEDV replicates in the small intestine and neonatal pigs rely on lactogenic immunity, it is important to develop a vaccine that can induce mucosal immunity via oral vaccination to induce antibodies in the lactogenic secretions of the sows (Zhou et al., 2019; Jang et al., 2023). Through the use of immunoinformatics approaches, large-scale processing for selecting suitable epitopes for vaccine construction would be faster and more cost-efficient than traditional vaccinology (Oyarzun et al., 2015). In summary, this study aimed to design an oral multiepitope vaccine against PEDV GIIa by targeting its S protein using immunoinformatics approaches. Materials and MethodsProteome retrieval and protein selectionOne hundred eight PEDV GIIa strains used in the study were referenced from the phylogenetic trees constructed by Yao, Qiao, et al. (2023), Yao, Zhu, et al. (2023), and Liu et al. (2022). Proteomes of each strain were retrieved from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/). Homologous sequences of the PEDV GIIa spike protein were then extracted from the proteomes using BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Multiple sequence alignment and masking variable residuesMultiple sequence alignment was performed on the extracted PEDV GIIa spike proteins using ClustalW in MEGA11 (Tamura et al., 2021). Variable Residues were masked using the Protein Variability Server (PVS) (http://imed.med.ucm.es/PVS/) with the variability threshold set to 1 (Diez-Rivero and Reche, 2010; Garcia-Boronat et al., 2008). The resulting protein sequence was subjected to Helper T Lymphocyte (HTL), Cytotoxic T Lymphocyte (CTL), and B cell epitope prediction. HTL epitope predictionThe NetMHCIIpan (https://services.healthtech.dtu.dk/services/NetMHCIIpan-4.0/) server was used to predict potential peptides with a strong binding affinity (<0.5) for 21 human leukocyte antigen (HLA) alleles (Reynisson et al., 2020). 15-mer peptides were generated, and their physicochemical properties were assessed. Antigenic, non-allergenic, and non-toxic peptides were further subjected to immunogenicity testing using IEDB CD4 immunogenicity prediction (http://tools.iedb.org/CD4episcore/) (Dhanda et al., 2018). All peptides with a combined score of >45 were immunogenic and counted as HTL epitopes. After identifying the final HTL epitopes, they were tested for their cytokine-inducing properties. Interferon-gamma (IFNγ)-inducing peptides were identified using the IFNepitope server (http://crdd.osdd.net/raghava/ifnepitope/). Peptides that were Interleukin-10 (IL-10) and Interleukin-4 (IL-4) inducers were identified using IL10Pred (https://webs.iiitd.edu.in/raghava/il10pred/index.html) (http://crdd.osdd.net/raghava/il4pred/) and IL4pred (http://crdd.osdd.net/raghava/il4pred/), respectively. Cytotoxic T lymphocyte (CTL) epitope predictionThe major histocompatibility complex (MHC) type I binding affinity was predicted using NetMHCcons 1.1 (https://services.healthtech.dtu.dk/services/NetMHCcons-1.1/) with a threshold of 0.5 for strong binders on 45 swine leukocyte antigen (SLA) alleles (Karosiene et al., 2012). 9-mer strong binding peptides were subjected to CTL epitope processing prediction using NetCTLpan 1.1 (https://services.healthtech.dtu.dk/services/NetCTLpan-1.1/) with the same alleles in the SLA supertype, a threshold for epitope identification of 1, 0.225 weight on C-terminal cleavage, and 0.025 weight on TAP efficiency (Stranzl, 2010). The physicochemical properties of the identified CTL epitopes were tested. After identifying the validated epitopes that were antigenic, non-allergenic, and non-toxic, a pMHCI immunogenicity test was performed. Class I Immunogenicity from IEDB (http://tools.iedb.org/immunogenicity/) was used for immunogenicity testing, and peptides with a score >0 were considered immunogenic and included as final CTL epitopes (Calis et al., 2013). Linear B lymphocyte (LBL) epitope predictionThe LBL epitopes were predicted using Bepipred (http://tools.iedb.org/bcell/) and verified using BcePred (http://crdd.osdd.net/raghava/bcepred/), ABCpred (https://webs.iiitd.edu.in/raghava/abcpred/), and SVMTriP (http://sysbio.unl.edu/SVMTriP/) to avoid server bias (Jespersen et al., 2017; Saha and Raghava, 2007; Saha and Raghava, 2006; Yao et al. 2012). The verified LBL epitopes were then tested for their physicochemical properties. Physicochemical properties assessment and immunogenicity predictionThe physicochemical evaluation of the epitopes include: 1) Antigenicity, 2) Toxicity, and 3) Allergenicity test. Antigenicity tests were first performed using VaxiJen v2.0 (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) (Doytchinova and Flower, 2007). All antigenic peptides were tested for their toxicity using ToxinPred (http://crdd.osdd.net/raghava/toxinpred/). Allergenicity tests were performed on all nontoxic peptides using AllerTOP v.2 (https://www.ddg-pharmfac.net/AllerTOP/) (Dimitrov et al., 2014). Vaccine constructionThe final epitopes were joined together using the following linkers: “KK” for LBL epitopes, “AAY” for CTL epitopes, and “GPGPG” for HTL epitopes. The linker “HEYGAEALERAG” was used to connect these epitope groups. Seven adjuvants were joined to the linked epitopes with the “EAAAK” linker to form seven different vaccine candidates. The adjuvants Sus scrofa β-defensin-1 (Fagbohun et al., 2022), F3-A6 ASFV hemagglutinin peptides (Argilaguet et al., 2012), phenol-soluble modulin α4 (Pang et al., 2022), 50S ribosomal protein L7/L12 (Maleki et al., 2022), heparin-binding hemagglutinin adhesin (HBHA) (Lei et al., 2020), cholera toxin B subunit (Lebens and Holmgren, 1994), and heat-labile enterotoxin IIc B chain precursor from Escherichia coli (Jobling, 2016) were attached to the N-terminus of the linked epitopes. The M-ligand sequences “CKSTHPLSC” and “CTGKSC” were added to the C-terminus of all assembled vaccine constructs. Fourteen (14) vaccine constructs were assembled in total. Physicochemical assessmentAll vaccine constructs were submitted to four different servers to assess their antigenicity, allergenicity, solubility, and stability. The following criteria were used in the selection of final vaccine constructs: (1) antigenicity score ≥ 0.40 in VaxiJen v2.0 (Doytchinova and Flower, 2007); (2) non-allergenic in AllerTOP v2.0 (Dimitrov et al., 2014); (3) soluble upon overexpression in E. coli in SCRATCH SolPro (https://scratch.proteomics.ics.uci.edu/) (Magnan et al., 2009); and (4) stable in Expasy Protparam (https://web.expasy.org/protparam/) (Gasteiger, 2003). Structure prediction and assessmentThe tertiary structure of the vaccine construct was predicted using ColabFold v1.5.2-patch (Mirdita et al., 2022) available in AlphaFold v.2 (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) (Jumper et al., 2021) and then refined using GalaxyRefine (https://galaxy.seoklab.org/cgi-bin/submit.cgi.type=REFINE). Refined models were selected based on the lowest Mol Probidity (Heo et al., 2013). In the case of similar Mol Probidity values, the models with the highest GDT-HA were selected. The local and overall model quality of the predicted structures was then assessed through Protein Structural Analysis (ProSA) (https://prosa.services.came.sbg.ac.at/) (Wiederstein and Sippl, 2007) and further validated by SAVES v6.0 (https://saves.mbi.ucla.edu/) ERRAT and PROCHECK (Agnihotry et al., 2022). The final selected structures met all of the following criteria: (1) z-score between −10 and 10 z-score in ProSa; (2) at least 95% overall quality factor in ERRAT; and (3) at least 90% favorable residues falling within the most favored regions in the PROCHECK Ramachandran plot. These were then visualized using ChimeraX (https://www.cgl.ucsf.edu/chimerax/) through an electrostatic molecule display (Goddard et al., 2017). Prediction of conformational B lymphocyte epitopesThe PDB files of the refined structures were used for discontinuous B-cell prediction in DiscoTope 3.0 (https://services.healthtech.dtu.dk/services/DiscoTope-3.0/). The epitope confidence threshold was set to higher confidence with the input structure type set to the AlphaFold structure. Immune simulationThe immune response profile of the final vaccine construct can be determined using the C-ImmSim server (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php?page=1). Three injections were administered at four-week intervals (time steps 1, 84, and 168). The random seed was set to 12345, the simulation volume to 50, and the simulation steps to 1000. The remaining parameters were set to default values. Molecular docking and molecular dynamicsMolecular interactions between the predicted vaccine construct and TLR4 were determined by molecular docking in Cluspro 2.0. The binding energy was determined using PRODIGY. PDBsum hin(https://www.ebi.ac.uk/thorntonsrv/databases/pdbsum/Generate.html) was used to determine the bonds between TLR4 and the PEDV vaccine constructs. The stability of the TLR4-Vaccine construct complex was analyzed through coarse grained (CG) molecular dynamics simulations in GROMACS 2023.2 using SIRAH force field in explicit solvent WatFour (WT4) (Darré et al., 2010). Two energy minimizations were done: 1) energy minimization of side chains by restraining the backbone, and 2) energy minimization of the whole system. Both were performed in 50,000 steps using the steepest descent algorithm (Machado et al., 2016). The system was then equilibrated for 5,000 ps at a temperature of 312 K and a pressure of 1 bar. To calculate the root mean square deviation (RMSD), the production simulation was run for 100 ns. Ethical approvalNot needed for this study. ResultsProteome retrieval and conservancy analysisOne-hundred-eight (108) spike (S) proteins were extracted from the proteomes of PEDV GIIa strains retrieved from Yao, Qiao, et al. (2023), Yao, Zhu, et al. (2023), and Liu et al. (2022) (Table 1). No variability was observed among the spike proteins at Shannon entropy (H) >1.0 in the PVS. Shannon entropy is a measure of the uncertainty in a given distribution (Carcassi et al., 2021). This implied a high degree of conservation of the S protein across the PEDV GIIa strain. Epitope predictionThe conserved sequence generated after PVS analysis was used to predict B and T cell epitopes. T lymphocytes are classified into cytotoxic and HTL. The activation of cytotoxic CD8+ T cells relies on peptide recognition of MHC class I proteins on the surface of antigen-presenting cells (APCs) (Alberts et al., 2002). Before the peptide is recognized by the T-cell receptor on CD8+ T-cells, the protein is first degraded by the proteosome and then transported by transporters associated with antigen processing (TAP) to the endoplasmic reticulum (ER). The peptide is further processed and then loaded onto MHC class I. Only short peptides 8 to 14 residues in length can bind to the MHC I cleft due to its closed end, with a preference for 9-mers, (Szeto et al., 2021; Tadros et al., 2023). β2-macroglobulin stabilizes the peptide-MHC class I complex. The complex is exported to the surface of the cell membrane for recognition by CD8 T cells in a process called licensing (Actor, 2019; Leichner, 2014). Following this pathway, the CTL epitope should be antigenic, cleaved by the proteasome, and efficiently transported by TAP. In this study, one hundred twelve peptides were predicted to have a high binding affinity to MHC I. MHC I processing analysis predicted 68 epitopes with efficient proteasomal cleavage, and efficient TAP transport. Of the 68 epitopes, 10 are antigenic, non-allergenic, non-toxic, and immunogenic. Listed in Table (2) are the candidate CTL epitopes and their corresponding physicochemical properties. During the activation of helper CD4+ T lymphocytes, the protein is broken down into peptides by proteases facilitated by the phagolysosome. The lysosome then fuses into MHC class II-containing vesicles where the peptides bind to MHC class II to be presented to the TCR of CD4+ (Leichner and Kambayashi, 2014). The binding groove of MHC class II proteins is open at both ends, allowing for the accommodation of the binding of longer peptides, 13–25 residues with a preference for 15-mers (Wieczorek et al., 2017; Tadros et al., 2022). The effector CD4+ can differentiate into different types depending on the cytokines they secrete (Alberts et al., 2014; Leichner and Kambayashi, 2014). Thus, a peptide is considered to be an HTL epitope when it can strongly bind to MHC II alleles and is antigenic, non-allergenic, non-toxic, and immunogenic. The cytokines secreted by the putative epitopes can then be analyzed to estimate their contribution during an immune response. One hundred fourty-four peptides were predicted to have a strong binding affinity to MHC II, with 97 peptides being antigenic. Of these, four were found to be non-allergenic, non-toxic, and immunogenic. The final list of HTL epitopes and their cytokine-inducing abilities are shown in Table (3). Unlike T-cell epitopes, the optimal length to induce maximum activity in B-cell epitopes has not been identified; hence, multiple servers are usually employed to increase prediction accuracy. A peptide is considered a candidate LBL epitope if it is predicted by two or more prediction servers (Galanis et al., 2021; Yang et al., 2021; Fontes et al., 2021). A total of 66 peptides were predicted by Bepipred 3.0, and validated by either BcePred, SVMTriP, or ABCpred. Six epitopes were found to be antigenic, non-allergenic, and non-toxic, and thus were considered as candidate LBL epitopes listed in Table (4). Vaccine construction and evaluationLBL, CTL, and HTL epitopes were connected using “KK,” “AAY,” and “GPGPG” linkers, respectively. The linker “HEYGAEALERAG” was used to connect the three different epitope groups with the “EAAAK” linker used to attach the adjuvant at the N-terminus of the sequence (Fig. 1). The final configuration of the vaccine construct is as follows: adjuvant - LBL-CTL-HTL, from the N- to the C-terminus. Seven adjuvants were used in this analysis: (1) Sus scrofa β-defensin-1, (2) F3-A6 ASFV hemagglutinin peptides, (3) phenol-soluble modulin α4, (4) 50S ribosomal protein L7/L12, (5) heparin-binding hemagglutinin adhesin (HBHA), (6) cholera toxin B subunit (CTB), and (7) heat-labile enterotoxin IIc B chain precursor (HIIc). To enhance the uptake of the identified epitopes to the M-cells, homing peptides were appended to the C-terminus of the constructs. Two homing peptides were utilized in this study with the following sequences “CKSTHPLSC” and “CTGKSC,” referred to in this study as M1 and M2, respectively. Overall, 14 candidate constructs were assembled and tested for antigenicity, allergenicity, stability, and solubility. All assembled vaccine constructs that were antigenic, non-allergenic, non-toxic, stable, and soluble were used for structure prediction (Table 5). Table 1. 108 PEDV GIIA strains as source for S protein sequences used in the study.
Table 2. Predicted CTL epitopes and their corresponding physicochemical properties.
Table 3. Predicted HTL epitopes and their corresponding physicochemical properties and cytokine-inducing abilities.
Table 4. Predicted LBL epitopes and their corresponding physicochemical properties.
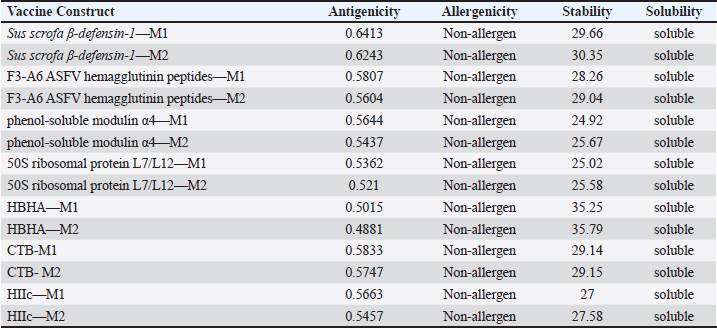
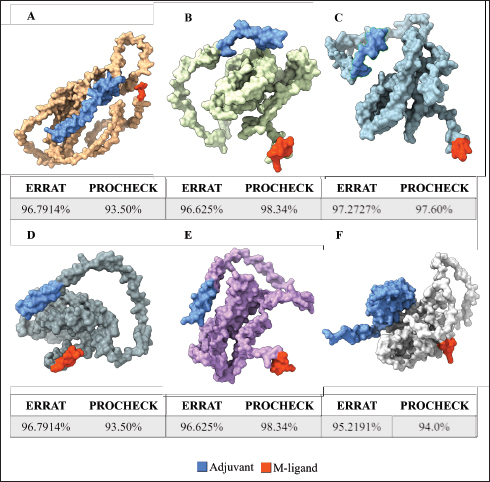
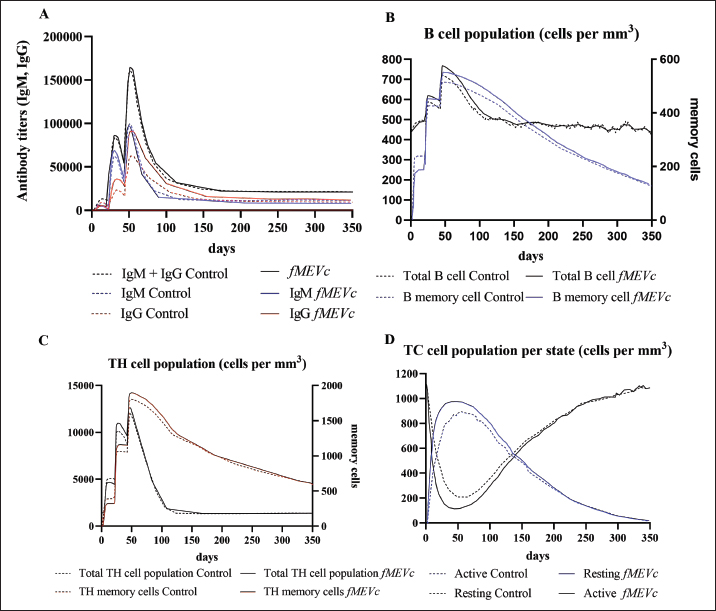
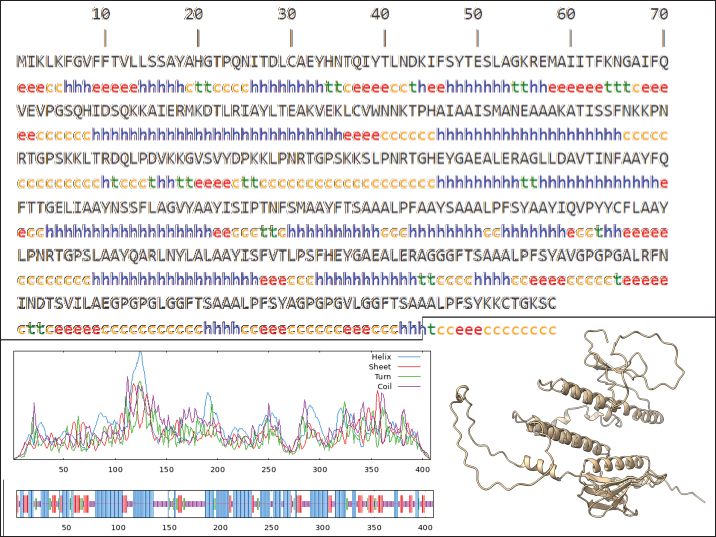
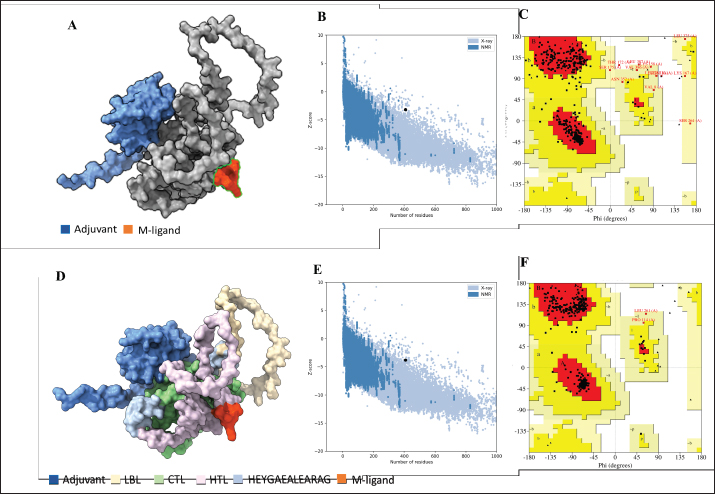
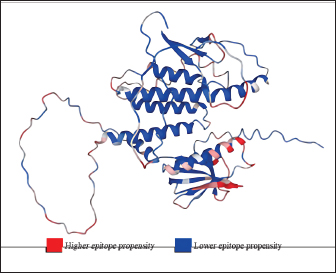
Fig. 1. General design of the final vaccine construct. Tertiary structure prediction and evaluationA tertiary structure is required to determine the molecular recognition of T-cell receptors (TCRs) (Greenbaum et al., 2007; Hou et al., 2023). An initial round of structure prediction and evaluation was performed to filter out candidate vaccine constructs. The protein sequences of the 15 vaccine constructs were used for structure prediction. Structures with an overall quality factor of 95% or greater in ERRAT, 90% of the residues or greater falling in the most favored residues in the Ramachandran plot in PROCHECK, and Z-score values falling within the Z-score range of similarly sized native proteins were considered to be of good quality. Six out of the fourteen vaccine constructs complied with all the threshold values and were thus considered of good quality (Fig. 2). Immune simulationThe immune simulation was performed to evaluate the host immune response post-immunization. There was a significant increase in the total antibody titers following the third immune response. Similarly, total TH cells and memory TH cells showed an increase in population after the first and second immune responses. There was also a steady increase in the TC cell population after immunization. IFN-γ was demonstrated to be the highest among the cytokines and interleukins induced. A comparison was made with a previous study that designed a PEDV multiepitope vaccine using the S proteins from four representative strains from each GII subgroup (Hou et al., 2023). Among the six candidates, one vaccine construct was shown to induce a more robust immune response compared to the previously designed PEDV vaccine by Hou et al. (2023) (Fig. 3). Structure prediction and evaluation of the final vaccine multiepitope vaccine construct (fMEVc)The fMEVc is 408 aa long and was predominantly composed of alpha helices (44.53%), followed by random coils (39.66%), and extended strains (15.82%) (Fig. 4). fMEVc has a theoretical pI of 9.26, instability index of 29.80, and a grand average of hydropathicity (GRAVY) value of 0.043, indicating that it is basic, stable upon overexpression, and hydrophobic. Antigenicity, solubility, and allergenicity tests showed that the fMEVc was safe (Table 6). The tertiary structure of fMEVc was generated using Alphafold. The generated structure had an ERRAT score of 87.8049% and z- score of −3.16. The Ramachandran plot showed that the initial structure had 83.6% residues falling within the most favored regions, 12.9% in additional allowed regions, 2.6% in generously allowed regions, and 3.7% in disallowed regions (Fig. 5A–C). To improve the quality of the structure, structural refinement was performed. The refined fMEVc structure had an ERRAT score of 95.2191% and z-score of −3.79, and the Ramachandran plot showed that 94.0% of the residues fell within the most favored regions, 5.7% in additional allowed regions, 0.3% in generally allowed regions, and none in disallowed regions, generating a structure with better quality (Fig. 5D–F). Conformational B cell epitope predictionAt a higher confidence level, the results showed that there were 30 residues distributed across 19 B-cell epitopes of fMEVc. The values range from 1.54517 to 4.57438. Figure 6 shows the identified conformational B cell epitopes with a deeper red color indicating a higher epitope propensity, while deeper blue-colored regions indicate a low epitope propensity. Table 5. Physicochemical properties of the sixteen (16) assembled vaccine constructs.
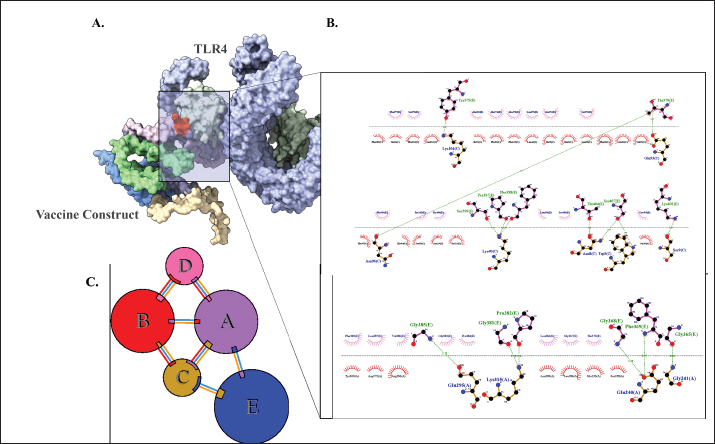
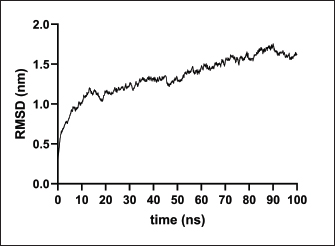
Fig. 2. Six good quality structures predicted and refined. A) Sus scrofa β-defensin—M2, B) F3-A6 ASFV hemagglutinin—M1, C) F3-A6 ASFV hemagglutinin—M2, D) phenolsoluble modulin α4—M1, E) phenol-soluble modulin α4—M2, and F) Cholera toxin B subunit—M2. Molecular docking and molecular dynamics of the final vaccine construct with fMEVcTLR4 activation results in innate immune system activation and, consequently, a more durable adaptive immune response (Sartorius et al., 2021). The final construct was docked using the ClusPro server. The TLR4-fMEVc complex had the highest binding affinity at a ΔG of −18.6 kcal/mol compared to the controls, TLR4-Brucella lumzaine synthase, and TLR4-M. tuberculosis RpfE and TLR4-S. pneumoniae DnaJ, with a binding energy of −9.9 kcal/mol, −9.7 kcal/mol, and −13.1 kcal/mol, respectively (Simbulan et al., 2024). Ligplot and PDBsum predicted 24 hydrogen bonds between the vaccine construct and TLR4 (Fig. 7). The stability of the vaccine construct-TLR4 complex was evaluated through coarse-grained MD simulations using the SIRAH force field (Dans et al., 2013). The RMSD over time plot was generated after the simulation, which analyzed the structural fluctuation of the overall structure of the vaccine construct-TLR4 complex. The simulation ran for 100ns with the protein complex stabilizing at approximately 75 ns. Thus, the vaccine construct was considered to be stable when bound to TLR4 (Fig. 8). DiscussionNeonatal pigs experience severe cases of PED, with mortality and morbidity rates reaching up to 100%, causing significant losses worldwide. This study targeted the spike (S) protein of PEDV since several studies have shown that the neutralizing activity is positively correlated to the IgA induced by the PEDV S protein (Song et al., 2016; Shan et al., 2022; Song et al., 2023). The S protein contains the S1 domain, which directs viral entry via binding to the host receptor, and the S2 domain, which deals with the merging of the virus to the host cell. Insertions, deletions, and substitutions that occur in the S protein during the evolution of the virus and are driven by genetic drift account for the diversity and relatedness of the virus. However, this could also strengthen or weaken the pathogenicity of PEDV, which makes it an essential indicator of virulence and a good target for vaccine development (Feng et al., 2022; Salamat et al., 2021; Zhuang et al., 2022).
Fig. 3. Immune simulation after immunization generated by C-immsim. A. Antibody titer population, B. B cell population, C. TH cell population, and D. TC cell population. A successful vaccine should be able to induce the adaptive immune system memory for fast immune recognition and response when exposed to the next antigen. There are two immune responses involved in the adaptive immune system: humoral immune responses facilitated by antibodies produced by B lymphocytes and a cellular immune response facilitated by T lymphocytes. (Yurina et al., 2022; Orosco, 2024b). This entails incorporating CTL, HTL, and LBL epitopes in the vaccine design. By including these epitopes, the vaccine aims to elicit a comprehensive immune response, involving both antibody-mediated defense and cellular immunity. T lymphocytes are classified into two main categories: CD8+ and CD4+ T cells. CD8+ T cells, cytotoxic T cells (CTLs), or killer T cells recognize MHC I-bound antigens. CD8+ T cells are naive and need to be activated to initiate effector functions. Effector CD8+ T cells migrate to the circulation and locate their antigenic targets expressed on infected cells. After its recognition, it begins its killing function. The killing action of CTLs can be one of two mechanisms: The first mechanism relies on the use of the Fas/Fas Ligand (FasL). FasL is expressed on activated CD8+ T cells and binds to the Fas (CD95) receptor found in many cell types. This activates caspases leading to apoptosis of the target cell. The other killing mechanism of CTL involves the release of two compounds that bypass the cell walls and active caspases, granzymes, and perforin (Moticka, 2016; Sauls et al., 2023). Conversely, CD4+ T cells or HTLs recognize MHC II-bound antigens. Similar to CD8+ T cells, CD4+ T cells start as naive and must undergo activation to initiate their effector actions. Effector CD4+ cells can differentiate into either Th1 or Th2 cells. Th1 cells secrete type I cytokines, such as IFN-y, while Th2 cells secrete type II cytokines, such as IL-4 and IL-5. Th17 cells secrete IL-17, which is important for phagocytic cell recruitment to inflammation sites. Th1 cells are important in combating intracellular pathogens by aiding cell-mediated responses and macrophage activation, Th2 is important during parasitic infection by aiding the humoral immune response, and Th17 cells are important during infection caused by extracellular bacteria (Alberts et al., 2014; Leichner and Kambayashi, 2014). Additionally, they activate the T cell-dependent part of humoral immunity, in which CD4+ recognizes antigens that usually elicit weak, if not absent, B cell responses (Sauls et al., 2023). T lymphocytes and B lymphocytes recognize epitopes that are short amino acid sequences that can induce an immune response and are thus essential in vaccine design (Zhang, 2019).
Fig. 4. The predicted secondary structure of the final vaccine construct. Table 6. Physicochemical properties of fMEVc.
Fig. 5. A. Predicted tertiary structure of the raw fMEVc and its corresponding B. ProsA and C. Ramachandran plot and D. refined fMEVc and its corresponding E. ProsA and F. Ramachandran plot.
Fig. 6. Predicted conformational B cell epitopes. Deeper red color indicates a higher epitope propensity while deeper blue color indicates lower epitope propensity. For CTL and HTL epitope prediction, this study utilized 45 SLA class I molecules and 21 HLA molecules from the original 27 HLA reference sets from IEDB because HLA class II displays strong homology to SLA class II. HLA-DP gene pairs were removed from the set because the SLA class II region does not contain these genes (Smith et al. 2005). These two reference sets are composed of alleles with the most frequently occurring specificities in the general population and represent commonly shared binding specificities (Martini, 2020). Ten CTL epitopes and four HTL epitopes were predicted to be antigenic, non-allergenic, non-toxic, and immunogenic, and were thus included in the final design. A CD8+ T cell-dominant CD4+/CD8+ ratio, as in this study, was expected to offer better protection against PEDV, as demonstrated by Annamalai et al. (2015). They observed an increase in CD4+ T cell frequencies in suckling pigs compared to weaned pigs, while no significant difference was observed in the frequencies of CD8+ T cells, indicating that neonates generally have polarity toward Th2 responses, implying the importance of inducing CD8+ T cells for PEDV infection.
Fig. 7. Molecular docking of vaccine construct with TLR4. A. Top cluster predicted by Cluspro of the docking of TLR4 (grey) with vaccine construct (colored) visualized using ChimeraX. B. Predicted interface residues by Ligplot in 2D. The green dotted lines indicate hydrogen bonds. C. Predicted interface residues by PDBsum in 2D. Chain E is the vaccine construct.
Fig. 8. RMSD analysis of fMEVC-TLR4 complex. In addition to the cellular response, the humoral response is an important component of the adaptive immune system. Specifically, antigen-antibodies are essential for the induction of a humoral response against invading pathogens. A specific antibody recognizes regions of an antigen called B cell epitopes (Potocnakova et al., 2016). Antibodies include neutralizing infectivity, antibody-dependent cellular cytotoxicity, phagocytosis, and complement-mediated lysis of infected cells or pathogens (Forthal, 2014). B cell epitopes can be either a continuous stretch of amino acids called linear epitopes or amino residues brought through protein folding, also called conformational epitopes (Forsström et al., 2015). This study identified six B linear B-cell epitopes and 13 key residues distributed across 19 B-cell epitopes. The predicted LBL. CTL and HTL epitopes were then linked using “KK,” “AAY,” and “GPGPG,” respectively. These linkers were added to enhance epitope presentation. “AAY” ensures the production of sites that will bind to the TAP transport, “GPGPG” facilitates the progression of immune cells, and “KK” facilitates the presentation of each peptide for the induction of antibodies. Each epitope group is then connected to each other using the “HEYGAEALERAG” which contains five cleavage sites important for eukaryotic proteosomal and lysososmal degradation systems (Andongma et al., 2023). Since epitope-based vaccines are usually associated with limited immunity, adjuvants are conjugated into the construct (Lei et al., 2019, Orosco and Espiritu 2024). The final vaccine design, fMEVc, is conjugated with Cholera toxin subunit B (CTB), which is the nontoxic binding component of cholera toxin. Its high immunogenicity and high binding affinity to membrane ganglioside GM1 on epithelial cells and fucosylated glycans, including microfold (M) cells, enterocytes, dendritic cells, and macrophages, indicate its potential to deliver bound antigens to mucosal APCs (Lavelle and Ward, 2022). This is particularly important during PEDV infection, as PEDV mainly replicates in porcine small intestinal villous epithelial cells, also known as enterocytes (Jang et al., 2023). To further ensure local induction of the mucosal immune response, the homing peptide “CTGKSC” was added. “CTGKSC” targets the M cells facilitating the delivery of the antigens from the luminal surface to the sub-epithelium which is important for maintaining mucosal immunity function (Andrieu et al., 2019; Dillon and Lo, 2019; Sauls and Taylor, 2022). The fMEVc was evaluated for its safety and was determined to be non-allergenic, non-toxic, and antigenic. Further physicochemical analysis showed that fMEVc was stable upon overexpression and was basic and slightly hydrophobic. Alphafold2 was employed for the prediction of the tertiary structure of fMEVc since a tertiary structure is required to determine the molecular recognition of T-cell receptors (TCRs) (Greenbaum et al., 2007; Hou et al., 2023). Alphafold2 ranked first in the CASP 14 ranking by a large margin (Kryshtafovych et al, 2021). The top predicted structure was then docked to TLR4. TLR4 recognizes envelope glycoproteins such as the spike glycoprotein (S) of PEDV (Kirchdoerfer et al., 2018; Saeng-chuto et al., 2022). Activation of TLR4, a transmembrane receptor, initiates intracellular signaling cascades, including MyD88-dependent and-independent pathways, thus establishing a protective immune response (Olejnik et al., 2018; Kim et al., 2023). fMEVc binds to the TLR4 with a binding energy of −18.0 kcal/mol, which is greater than the binding energy of the controls suggesting that the fMEVc is able to activate TLR4. Furthermore, a molecular dynamics simulation was carried out to assess the stability of the dock. RMSD analysis, which measures the deviation of the final structure from the initial structure, was performed (GROMACS 2024). The results of the simulation showed that the fMEVc-TLR4 complex stabilized at 75 ns, further proving the ability of fMEVc to activate TLR4. To predict the in vitro response of fMEVc, an immune simulation using C-immsim was implemented. Three fMEVc immunizations were conducted every 7 days, as described by Hou et al. (2023). As previously mentioned, the protection of neonatal pigs against PEDV relies on lactogenic immunity. IgG abundance in the colostrum is one way to achieve lactogenic immunity. fMEVc showed an increase in IgG after three immunizations, as well as the induction of memory B cells. Both groups showed greater values than those in the control group. This is especially notable because numerous studies have underscored the significance of inducing neutralizing antibodies against PEDV, specifically in the intestinal mucosa (Langel et al., 2019; Jang et al., 2023; Song et al., 2023; Lei et al., 2024). With the inclusion of a mucosal adjuvant, a homing peptide, and abundant production of memory B cells, TH cells, and TC cells, fMEVc has the potential to successfully induce a robust mucosal immune response. ConclusionIn conclusion, the development of an efficacious vaccine against PEDV remains crucial for mitigating its devastating impact on neonatal pig populations worldwide. Current vaccines have limitations in inducing sufficient immune responses, particularly against emerging strains. To circumvent this problem, the current vaccine design in this study, fMEVc, integrates predicted conserved CTL, HTL, and B cell epitopes across 108 PEDV-GIIa spike protein sequences coupled with CTB for enhanced mucosal delivery and immunogenicity. Physicochemical analyses confirmed its safety and stability, while molecular docking studies have demonstrated its potential to activate TLR4, an essential component of the innate immune response. Immune simulations predicted a robust response characterized by increased IgG levels and memory B cell induction, underscoring the promise of fMEVc in providing effective protection against PEDV. However, similar to most in silico studies, the findings of this study should be further validated in wet laboratory experiments using model organisms. AcknowledgmentsThe authors would like to thank Mr. John Christian de Guzman, Mr. Albert Neil Dulay, and Dr. Mark Andrian Macalalad for their invaluable assistance in conducting the molecular dynamics simulations performed in this study. Conflict of interestThe authors declare that there is no conflict of interest. Author contributionsFLO conceptualized and acquired funding for the study. EMJSS, ECB, NMOO, and LEF conceptualized and optimized the workflow in the paper. EMJSS, FFF, HABR, and APAD conducted the analysis and drafted the paper. EMJSS, ECB, NMOO, LEF, and FLO revised the paper. All authors approved the final version of the manuscript for submission. FundingThe authors would like to express their gratitude to the Philippine Council for Agriculture, Aquatic and Natural Resources Research and Development (DOST-PCAARRD) for funding this research project under the Virology and Vaccine Research and Development Program (VVRDP). Data availabilityThe main data supporting the findings of this study are available within the manuscript. Additional supporting data are available from the authors upon reasonable request. ReferencesActor, J.K. 2019. Chapter 4—T Lymphocytes: ringleaders of adaptive immune function. In introductory immunology (Second Edition). Ed., Actor, J.K. Cambridge, MA: Academic Press, pp: 45–62. Agnihotry, S., Pathak, R.K., Singh, D.B., Tiwari, A. and Hussain, I. 2022. Chapter 11—Protein structure prediction. In Bioinformatics. Eds., Singh, D.B., Pathak, R.K. Cambridge, MA: Academic Press, pp: 177–188. Alberts, B., Johnson, A., Lewis, J., Raff, M., Roberts, K. and Walter, P. 2002. T Cells and MHC proteins.in molecular biology of the cell. 4th edition. New York, NY: Garland Science. Andrieu, J., Re, F., Russo, L. and Nicotra, F. 2019. Phage-displayed peptides targeting specific tissues and organs. J. Drug. Targeting. 27, 555–565. Annamalai, T., Saif, L. J., Lu, Z. and Jung, K. 2015. Age-dependent variation in innate immune responses to porcine epidemic diarrhea virus infection in suckling versus weaned pigs. Vet. Immunol. Immunopathol. 168, 193–202. Calis, J.J.A., Maybeno, M., Greenbaum, J.A., Weiskopf, D., Silva, A.D.D., Sette, A., Keşmir, C. and Peters, B. 2013. Properties of MHC class I presented peptides that enhance immunogenicity. PLOS Comput. Biol. 9, e1003266. Carcassi, G., Aidala, C.A. and Barbour, J. 2021. Variability as a better characterization of Shannon entropy. Eur. J. Phys. 42, 045102. Dans, P.D., Darré, L., Machado, M.R., Zeida, A., Brandner, A.F. and Pantano, S. 2013. Assessing the accuracy of the SIRAH force field to model DNA at coarse grain level. In advances in bioinformatics and computational biology. Eds. Setubal, J.C., Almeida, N.F. Lecture notes in computer science. Springer International Publishing, Recife, Brazil, pp: 71–81. Darré, L., Machado, M.R., Dans, P.D., Herrera, F.E. and Pantano, S. 2010. Another coarse grain model for aqueous solvation: WAT FOUR? J. Chem. Theory Comput. 6, 3793–3807. Diez Rivero, C. and Reche, P.A. 2010. Discovery of conserved epitopes through sequence variability analyses. Springer. Dillon, A. and Lo, D. D. 2019. M Cells: intelligent engineering of mucosal immune surveillance. Front. Immunol. 10. Dimitrov, I., Bangov, I., Flower, D.R. and Doytchinova, I. 2014. AllerTOP v.2—a server for in silico prediction of allergens. J. Mol. Model. 20, 2278. Doytchinova, I. A. and Flower, D. R. 2007. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8, 4. Feng, B., Li, C., Qiu, Y., Qi, W., Qiu, M., Li, J., Lin, H., Zheng, W., Zhu, J. and Chen, N. 2023. Genomic characterizations of porcine epidemic diarrhea viruses (PEDV) in diarrheic piglets and clinically healthy adult pigs from 2019 to 2022 in China. Animals 13, 1562. Fontes, S. da S., Maia, F. de M., Ataides, L.S., Conte, F.P., Lima-Junior, J. da C., Rozental, T., da Silva Assis, M.R., Júnior, A.A.P., Fernandes, J., de Lemos, E.R.S. and Rodrigues-da-Silva, R.N. 2021. Identification of immunogenic linear B-cell epitopes in C. burnetii outer membrane proteins using immunoinformatics approaches reveals potential targets of persistent infections. Pathogens 10, 1250. Forsström, B., Bisławska Axnäs, B., Rockberg, J., Danielsson, H., Bohlin, A. and Uhlen, M. 2015. Dissecting antibodies with regards to linear and conformational epitopes. PLoS One 10, e0121673. Forthal, D.N. 2014. Functions of antibodies. Microbiol Spectr. 2, 1–17. Garcia-Boronat, M., Diez-Rivero, C. M., Reinherz, E.L. and Reche, P.A. 2008. PVS: a web server for protein sequence variability analysis tuned to facilitate conserved epitope discovery. Nucleic Acids Res. 36, W35–W41. Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R.D. and Bairoch, A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788. Goddard, T.D., Huang, C.C., Meng, E.C., Pettersen, E.F., Couch, G.S., Morris, J.H. and Ferrin, T.E. 2018. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. Greenbaum, J.A., Andersen, P.H., Blythe, M., Bui, H.H., Cachau, R.E., Crowe, J., Davies, M., Kolaskar, A.S., Lund, O., Morrison, S., Mumey, B., Ofran, Y., Pellequer, J.L., Pinilla, C., Ponomarenko, J.V., Raghava, G.P.S., van Regenmortel, M.H. V., Roggen, E.L., Sette, A., Schlessinger, A., Sollner, J., Zand, M. and Peters, B. 2007. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J. Mol. Recognit. 20, 75–82. Heo, L., Park, H. and Seok, C. 2013. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41, W384–W388. Hou, W., Wu, H., Wang, S., Wang, W., Wang, B. and Wang, H. 2023. Designing a multi-epitope vaccine to control porcine epidemic diarrhea virus infection using immunoinformatics approaches. Front. Microbiol. 14, 1264612. IJMS | Free full-text | Linear B-Cell epitope prediction for in silico vaccine design: a Performance Review of Methods Available via Command-Line Interface. Jang, G., Lee, D., Shin, S., Lim, J., Won, H., Eo, Y., Kim, C.-H. and Lee, C. 2023. Porcine epidemic diarrhea virus: an update overview of virus epidemiology, vaccines, and control strategies in South Korea. J. Vet. Sci. 24, e58. Jespersen, M.C., Peters, B., Nielsen, M. and Marcatili, P. 2017. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 45, W24–W29. Jobling, M. G. 2016. The chromosomal nature of LT-II enterotoxins solved: a lambdoid prophage encodes both LT-II and one of two novel pertussis-toxin-like toxin family members in type II enterotoxigenic Escherichia coli. Pathogens Dis. 74, ftw001. Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S.A.A., Ballard, A.J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., Back, T., Petersen, S., Reiman, D., Clancy, E., Zielinski, M., Steinegger, M., Pacholska, M., Berghammer, T., Bodenstein, S., Silver, D., Vinyals, O., Senior, A.W., Kavukcuoglu, K., Kohli, P. and Hassabis, D. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. Karosiene, E., Lundegaard, C., Lund, O. and Nielsen, M. 2012. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics 64, 177–186. Kim, H.J., Kim, H., Lee, J.H. and Hwangbo, C. 2023. Toll-like receptor 4 (TLR4): new insight immune and aging. Immun. Ageing. 20, 67. Kirchdoerfer, R.N., Wang, N., Pallesen, J., Wrapp, D., Turner, H.L., Cottrell, C.A., Corbett, K.S., Graham, B.S., McLellan, J.S. and Ward, A.B. 2018. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci. Rep. 8, 15701. Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K. and Moult, J. 2021. Critical assessment of methods of protein structure prediction (CASP)—Round XIV. Proteins Struct. Funct. Bioinform. 89, 1607–1617. Lavelle, E.C. and Ward, R.W. 2022. Mucosal vaccines — fortifying the frontiers. Nat. Rev. Immunol. 22, 236–250. Lebens, M. and Holmgren, J. 1994. Structure and arrangement of the cholera toxin genes in Vibrio cholerae O139. FEMS Microbiol. Lett. 117, 197–202. Lei, Y., Shao, J., Ma, F., Lei, C., Chang, H. and Zhang, Y. 2020. Enhanced efficacy of a multi-epitope vaccine for type A and O footand-mouth disease virus by fusing multiple epitopes with Mycobacterium tuberculosis heparin-binding hemagglutinin (HBHA), a novel TLR4 agonist. Mol. Immunol. 121, 118–126. Lei, Y., Zhao, F., Shao, J., Li, Y., Li, S., Chang, H. and Zhang, Y. 2019. Application of built-in adjuvants for epitope-based vaccines. PeerJ. 6, e6185. Leichner, T. and Kambayashi, T. 2014. White blood cells and lymphoid tissue.in reference module in biomedical Ssiences. Amsterdam, The Netherlands: Elsevier. Liu, H., Yin, X., Tian, H., Qiu, Y., Wang, Z., Chen, J., Ma, D., Zhao, B., Du, Q., Tong, D. and Huang, Y. 2022. The S protein of a novel recombinant PEDV strain promotes the infectivity and pathogenicity of PEDV in mid-west China. Transbound. Emerg. Dis. 69, 3704–3723. Liu, X., Zhang, L., Zhang, Q., Zhou, P., Fang, Y., Zhao, D., Feng, J., Li, W., Zhang, Y. and Wang, Y. 2019. Evaluation and comparison of immunogenicity and cross-protective efficacy of two inactivated cell culture-derived GIIa- and GIIb-genotype porcine epidemic diarrhea virus vaccines in suckling piglets. Vet. Microbiol. 230, 278–282. Machado, M.R. and Pantano, S. 2016. SIRAH tools: mapping, backmapping and visualization of coarse-grained models. Bioinform. 32, 1568–1570. Magnan, C.N., Randall, A. and Baldi, P. 2009. SOLpro: accurate sequence-based prediction of protein solubility. Bioinform. 25, 2200–2207. Maleki, A., Russo, G., Parasiliti Palumbo, G.A. and Pappalardo, F. 2022. In silico design of recombinant multi-epitope vaccine against influenza A virus. BMC Bioinform. 22, 617. Martini, S. 2020. HLA allele frequencies and reference sets with maximal population coverage. IEDB Solutions Center. Morales, R.G., Umandal, A.C. and Lantican, C.A. (2007, November). Emerging and reemerging diseases in Asia and the Pacific with special emphasis on porcine epidemic diarrhea. In Conference OIE, Paris, France, pp: 185–189. Olejnik, J., Hume, A.J. and Mühlberger, E. 2018. Toll-like receptor 4 in acute viral infection: too much of a good thing. PLoS Pathogens 14, e1007390. Orosco, F.L. 2024a. Immune evasion mechanisms of porcine epidemic diarrhea virus: a comprehensive review. Vet. Integ. Sci. 22, 171–192. Orosco, F.L. 2024b. Recent advances in peptide-based nanovaccines for re-emerging and emerging infectious diseases. J. Adv. Biotechnol. Exp. Ther. 7, 106. Orosco, F.L. and Espiritu, L.M. 2024. Navigating the landscape of adjuvants for subunit vaccines: recent advances and future perspectives. Int. J. App. Pharm. 18–32. Oyarzun, P., Ellis, J.J., Gonzalez-Galarza, F.F., Jones, A.R., Middleton, D., Boden, M. and Kobe, B. 2015. A bioinformatics tool for epitope-based vaccine design that accounts for human ethnic diversity: application to emerging infectious diseases. Vaccine 33, 1267–1273. Pang, M., Tu, T., Wang, Y., Zhang, P., Ren, M., Yao, X., Luo, Y. and Yang, Z. 2022. Design of a multi-epitope vaccine against Haemophilus parasuis based on pan-genome and immunoinformatics approaches. Front. Vet. Sci. 9, 1053198. Paraguison-Alili, R. and Domingo, C.Y.J. 2016. Phylogenetic tracking of current porcine epidemic diarrhea virus (PEDV) strains in the Philippines. Arch. Virol. 161, 2601–2604. Potocnakova, L., Bhide, M. and Pulzova, L.B. 2016. An introduction to B-cell epitope mapping and in silico epitope prediction. J. Immunol. Res. 2016, 6760830. Reynisson, B., Barra, C., Kaabinejadian, S., Hildebrand, W.H., Peters, B. and Nielsen, M. 2020. Improved prediction of MHC II antigen presentation through integration and motif deconvolution of mass spectrometry MHC eluted ligand data. J. Proteome Res. 19, 2304–2315. Saeng-chuto, K., Madapong, A., Kaeoket, K., Piñeyro, P.E., Tantituvanont, A. and Nilubol, D. 2022. Co-infection of porcine deltacoronavirus and porcine epidemic diarrhea virus induces early TRAF6-mediated NF-κB and IRF7 signaling pathways through TLRs. Sci. Rep. 12, 19443. Saha, S. and Raghava, G.P.S. 2006. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Protein Struct. Funct. Bioinform. 65, 40–48. Saha, S. and Raghava, G.P.S. 2007. Prediction methods for B-cell epitopes. In immunoinformatics: predicting immunogenicity in silico. Ed., Flower. Methods in Molecular BiologyTM. Totowa, NJ: Humana Press, pp: 387–394. Salamat, S.E.A., Collantes, T.M.A., Lumbera, W.M.L., Tablizo, F.A., Mutia, C.T.M., Ong, J.D.P. and Bandoy, D.J.D.R. 2021. Sequence analysis of new variants of porcine epidemic diarrhea virus in Luzon, Philippines, in 2017. Arch. Virol. 166, 1859–1867. Sauls, R.S. and Taylor, B.N. 2022. Histology, M Cell.in StatPearls. Treasure Island, FL: StatPearls Publishing. Simbulan, A.M., Banico, E.C., Sira, E.M.J.S., Odchimar, N.M.O. and Orosco, F.L. 2024. Immunoinformatics-guided approach for designing a pan-proteome multi-epitope subunit vaccine against African swine fever virus. Sci. Rep. 14, 1354. Smith, D.M., Lunney, J.K., Ho, C.S., Martens, G.W., Ando, A., Lee, J.H., Schook, L., Renard, C. and Chardon, P. 2005. Nomenclature for factors of the swine leukocyte antigen class II system, 2005. Tissue Antigens 66, 623–639. Stranzl, T., Larsen, M. V., Lundegaard, C. and Nielsen, M. 2010. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62, 357–368. Szeto, C., Lobos, C.A., Nguyen, A.T. and Gras, S. 2021. TCR recognition of peptide–MHC-I: rule makers and breakers. Int. J. Mol. Sci. 22, 68. Tadros, D.M., Eggenschwiler, S., Racle, J. and Gfeller, D. 2023. The MHC Motif Atlas: a database of MHC binding specificities and ligands. Nucleic Acids Res. 51, D428–D437. Tamura, K., Stecher, G. and Kumar, S. 2021. MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. Wieczorek, M., Abualrous, E.T., Sticht, J., Álvaro-Benito, M., Stolzenberg, S., Noé, F. and Freund, C. 2017. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front. Immunol. 8, 292. Wiederstein, M. and Sippl, M.J. 2007. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 35, W407–W410. Yang, Z., Bogdan, P. and Nazarian, S. 2021. An in silico deep learning approach to multi-epitope vaccine design: a SARS-CoV-2 case study. Sci. Rep. 11, 3238. Yao, X., Qiao, W.T., Zhang, Y.Q., Lu, W.H., Wang, Z.W., Li, H.X. and Li, J.L. 2023a. A new PEDV strain CH/HLJJS/2022 can challenge current detection methods and vaccines. Virol. J. 20, 13. Yao, B., Zhang, L., Liang, S. and Zhang, C. 2012. SVMTriP: A Method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS One 7, e45152. Yao, X., Zhu, Y., Qiao, W.T., Lu, W.H., Zhang, Y.Q. and Li, J.L. 2023b. Based on the results of PEDV phylogenetic analysis of the most recent isolates in China, the occurrence of further mutations in the antigenic site S1° and COE of the S protein which is the target protein of the vaccine. Transbound. Emerg. Dis. 2023, e1227110. Yu, K., Liu, X., Lu, Y., Long, M., Bai, J., Qin, Q., Su, X., He, G., Mi, X., Yang, C., Wang, R., Wang, H., Chen, Y., Wei, Z., Huang, W. and Ouyang, K. 2023. Biological characteristics and pathogenicity analysis of a low virulence G2a porcine epidemic diarrhea virus. Microbiol. Spectrum 11, e04535-22. Yurina, V. and Adianingsih, O.R. 2022. Predicting epitopes for vaccine development using bioinformatics tools. Ther. Adv. Vaccines Immunother. 10, 25151355221100218. Zhang, G., Keskin, D.B. and Chitkushev, L. 2019. Extraction of immune epitope information. In encyclopedia of bioinformatics and computational biology - Eds., Ranganathan, S., Gribskov, M., Nakai, K., Schönbach, C. Elsevier, Amsterdam, Netherlands. Zhou, Y., Chen, C., Chen, Y., Liu, Z., Zheng, J., Wang, T., Luo, H., Liu, Y., Shan, Y., Fang, W. and Li, X. 2019. Effect of route of inoculation on innate and adaptive immune responses to porcine epidemic diarrhea virus infection in suckling pigs. Vet. Microbiol. 228, 83–92. Zhuang, H., Sun, L., Wang, X., Xiao, M., Zeng, L., Wang, H., Yang, H., Lin, F., Wang, C., Qin, L. and Wang, C. 2022. Molecular characterization and phylogenetic analysis of porcine epidemic diarrhea virus strains circulating in China from 2020 to 2021. BMC Vet. Res. 18, 392. | ||

| How to Cite this Article |
| Pubmed Style Sira EMJS, Banico EC, Odchimar NMO, Fajardo LE, Jr. FFF, Refuerzo HAB, Dictado APA, Orosco FL. Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein. Open Vet. J.. 2024; 14(5): 1224-1242. doi:10.5455/OVJ.2024.v14.i5.18 Web Style Sira EMJS, Banico EC, Odchimar NMO, Fajardo LE, Jr. FFF, Refuerzo HAB, Dictado APA, Orosco FL. Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein. https://www.openveterinaryjournal.com/?mno=194064 [Access: January 25, 2026]. doi:10.5455/OVJ.2024.v14.i5.18 AMA (American Medical Association) Style Sira EMJS, Banico EC, Odchimar NMO, Fajardo LE, Jr. FFF, Refuerzo HAB, Dictado APA, Orosco FL. Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein. Open Vet. J.. 2024; 14(5): 1224-1242. doi:10.5455/OVJ.2024.v14.i5.18 Vancouver/ICMJE Style Sira EMJS, Banico EC, Odchimar NMO, Fajardo LE, Jr. FFF, Refuerzo HAB, Dictado APA, Orosco FL. Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein. Open Vet. J.. (2024), [cited January 25, 2026]; 14(5): 1224-1242. doi:10.5455/OVJ.2024.v14.i5.18 Harvard Style Sira, E. M. J. S., Banico, . E. C., Odchimar, . N. M. O., Fajardo, . L. E., Jr., . F. F. F., Refuerzo, . H. A. B., Dictado, . A. P. A. & Orosco, . F. L. (2024) Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein. Open Vet. J., 14 (5), 1224-1242. doi:10.5455/OVJ.2024.v14.i5.18 Turabian Style Sira, Ella Mae Joy Sinco, Edward Coralde Banico, Nyzar Mabeth Obemio Odchimar, Lauren Emily Fajardo, Ferdinand Fajutagana Fremista Jr., Hanna Angelika Bobis Refuerzo, Ana Patrisha Aribon Dictado, and Fredmoore Legaspi Orosco. 2024. Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein. Open Veterinary Journal, 14 (5), 1224-1242. doi:10.5455/OVJ.2024.v14.i5.18 Chicago Style Sira, Ella Mae Joy Sinco, Edward Coralde Banico, Nyzar Mabeth Obemio Odchimar, Lauren Emily Fajardo, Ferdinand Fajutagana Fremista Jr., Hanna Angelika Bobis Refuerzo, Ana Patrisha Aribon Dictado, and Fredmoore Legaspi Orosco. "Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein." Open Veterinary Journal 14 (2024), 1224-1242. doi:10.5455/OVJ.2024.v14.i5.18 MLA (The Modern Language Association) Style Sira, Ella Mae Joy Sinco, Edward Coralde Banico, Nyzar Mabeth Obemio Odchimar, Lauren Emily Fajardo, Ferdinand Fajutagana Fremista Jr., Hanna Angelika Bobis Refuerzo, Ana Patrisha Aribon Dictado, and Fredmoore Legaspi Orosco. "Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein." Open Veterinary Journal 14.5 (2024), 1224-1242. Print. doi:10.5455/OVJ.2024.v14.i5.18 APA (American Psychological Association) Style Sira, E. M. J. S., Banico, . E. C., Odchimar, . N. M. O., Fajardo, . L. E., Jr., . F. F. F., Refuerzo, . H. A. B., Dictado, . A. P. A. & Orosco, . F. L. (2024) Immunoinformatics approach for designing a multi-epitope subunit vaccine against porcine epidemic diarrhea virus genotype IIA spike protein. Open Veterinary Journal, 14 (5), 1224-1242. doi:10.5455/OVJ.2024.v14.i5.18 |

