




| Original Article | ||
Open Vet J. 2022; 12(1): 61-68 Open Veterinary Journal, (2022), Vol. 12(1): 61–68 Original Research Metagenomic analysis of the intestinal microbiome in goats on cactus and Salicornia-based dietsFredy Fabián Domínguez1,2*, Milly Edith Vega Crisanto3, Rosa Liliana Solís Castro3, Lourdes Vásquez Rojas1, Vanessa Miluska Baylon Cuba1, Gabriela Raquel Sucapuca Santos1, Carlos Alberto Luque Ramos1 and Eric Mialhe41Public Technological Higher Education Institute “July 24, Zarumilla,” Tumbes, Perú 2Agrarian Sciences Faculty, National University of San Martin, Tarapoto, Perú 3Health Sciences Faculty, National University of Tumbes, Tumbes, Perú 4Molecular Biotechnology Laboratory, INCABIOTEC, Tumbes, Perú *Corresponding Author: Fredy Fabián Domínguez. Agrarian Sciences Faculty, National University of San Martin, Tarapoto, Perú. Tel.: + 51 962644603. Email: ffabian [at] unsm.edu.pe Submitted: 10/10/2021 Accepted: 04/01/2022 Published: 23/01/2022 © 2022 Open Veterinary Journal
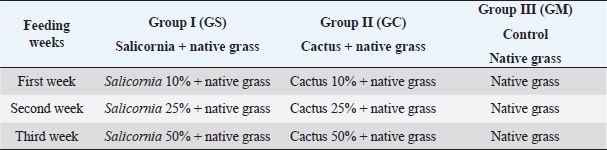
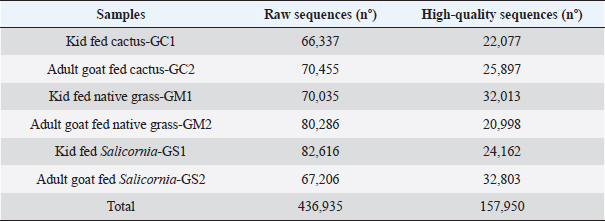
AbstractBackground: The Peruvian coast is characterized by its arid and saline soils, the cactus being an alternative for arid soils and Salicornia for saline soils. Therefore, it is necessary to develop nutrition based on the intestinal microbiota in goats. Aim: To identify the intestinal microbiota in goats through a metagenomic analysis. Methods: In this study, goats and kids were randomly selected and fed cacti and Salicornia as potential forage species compared to native grass to study the changes in the microbiota using massive sequencing using the 16S rRNA gene as a marker. Results: The sequencing results showed the taxonomic levels of Bacteroidetes and Firmicutes at the phylum level as the most abundant in creole goats’ microbiome, varying from 18% to 36% and 47% to 66%, respectively. At the genus level, variants of the genus Ruminococcaceae stand out, related to cellulose degradation, as the most dominant in all samples, followed by Christensenellaceae, Rikenellaceae, and Prevotellaceae. Also, the genus Akkermansia appeared in greater abundance in kids fed with cactus, being necessary for being related to the intestinal mucosa’s health and avoiding the adhesion of pathogens to the intestinal epithelium. Conclusion: These microbiota changes based on diets with high fiber content are necessary to understand the adaptation of this species to favorable dietary changes. Keywords: Microbiota, Forage, Ruminants, Bacteria, Bioinformatic. IntroductionThe diverse and complex symbiotic microbial community that houses the intestinal tract contributes significantly to the health and disease of mammals. These microbes, led in a higher proportion by bacteria, play a crucial role in the host’s nutrition, physiology, behavior, metabolic, and regulatory networks (McFall-Ngai et al., 2013). Many of these microbes are specifically inherited and unchanged over time through a maternal transfer to the infant (Funkhouser and Bordenstain et al., 2013), and which in turn, from birth, is rapidly colonized by a microbial community (Frese et al., 2015), and in its subsequent development it changes depending on the genetics, environmental variables, and diets of the host (Benson et al., 2010). In diets, such as the diet based on plant fibers (pectin, cellulose, and xylan) considered the primary natural food source, it models the shape and performance of the diverse microbial community in ruminants, generating carbohydrates, short-chain fatty acids, and proteins that are absorbed through the different areas of the gastrointestinal organ (Jiang et al., 2020), which are essential for its growth, reproduction, thermoregulation, and immunity of the host. For many years, studying this microbiota in ruminants has been vital to improving their production (Egan, 2005). These studies were based on traditional techniques through routine enzymatic assays to quantify cellulase, xylanase, and proteolytic activity (McSweeney et al., 2005). They managed to isolate several species that, at the time, were only related to digestion and pathogenicity. Today, with the biotechnological advances in massive DNA sequencing, an unprecedented number of researches related to microorganisms have opened, especially in noncultivable ones. These initiatives have generated massive amounts of publicly available data that have significantly increased the depth and breadth of knowledge of the gut microbiome in many species and environments (Allali et al., 2017). In Perú, the production of creole goats on the northern coast stands out for their ability to withstand long months of drought, with good fertility and milk production traits. However, traditional breeders’ inadequate technical preparation preserves this activity with a precarious diet based on harvest residues, natural forages, and seasonal shrub species from dry forests, thus limiting their development and production (Arroyo, 1998). Therefore, new breeding methods, health, and livestock feeding are urgently needed to improve the producers’ economic resources and living conditions. A great alternative to increasing goats’ livelihood in critical periods is by incorporating forage species from arid zones such as succulents into their nutrition. With a high prevalence in northern Perú, these plants have high nutritional values that allow reaching the productive levels necessary for goat development (Guevara and Estévez, 2001). In this study, young and adult goats were fed with cacti and Salicornia as potential forage species compared to native grass to study the changes in the microbiota using massive sequencing using the 16S rRNA gene as a marker. Materials and MethodsSample collectionThe research was carried out on the farm of the Instituto de Educación Superior Tecnológico Público 24 de Julio, Zarumilla, Tumbes, Perú. The experimental specimens of similar age and weight were randomly selected from 30 adult goats (approx. 4 years) and 30 young goats (3 months). The study was divided into three groups: group I (GS) formed by two animals (adult goat and kid) and fed with Salicornia; Group II (GC) also consisting of the same set up as the former and fed with cactus; and the control group (GM) fed with local natural forage (Table 1) to evaluate the effect of these diets on the intestinal microbiota, to develop an efficient nutrient plan for the animals. Extraction of DNAThe samples were obtained 21 days after the end of the investigation. The fecal samples were taken directly from the anus of each of the specimens belonging to each treatment and were taken to the laboratory to be stored at 4°C for their respective processing. The extraction of genomic DNA from the samples was carried out using the Quick-DNATM Fecal/Soil Microbe Miniprep Kit following the supplier’s steps. The quality of the extracted DNA was carried out by 1.5% agarose gel electrophoresis (Huang et al., 2018). The metagenomic DNA samples obtained were stored at −20°C. Sequencing and bioinformatics analysisSamples were amplified by polymerase chain reaction using the HotStarTaq Master Plus Mix Kit (Qiagen, USA). The amplification conditions were as follows: 94°C for 3 minutes, followed by 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds, and 72°C for 1 minute; elongation at 72°C for 5 minutes; and the sequencing was carried out using the Ion Torrent sequencing platform PGM on the variable region V4 16S rRNA using primers 515f/806r (Caporaso et al., 2011) by the company MR DNA (www.mrdnalab.com, Shallowater, TX). The sequences generated by Ion Torrent PGM were processed and analyzed using the bioinformatics software Quantitative Insights Into Microbial Ecology (QIIME; http://qiime.org) version 1.9.1 (Caporaso et al., 2011). Then, the sequences were filtered with quality scores > Q 25, length > 150 base pairs, and with UCHIME chimera sequences (Edgar et al., 2011). The non-chimera sequences were assigned in operational taxonomic units (OTUs) with 97% taxonomic identity to the SILVA v128 database (https://www.arb-silva.de/) for the taxonomic identification of bacteria. Finally, OTUs less than 0.05% were filtered (Bokulich et al., 2013). Each sample calculated the alpha diversity indices with richness (Chao1) and diversity (Shannon–Wiener and Simpson). Together, the beta diversity indices were used to differentiate the bacterial communities’ structure in each sample using QIIME and Phyloseq and vegan packages in RStudio v. 3.6.2. (https://www.rstudio.com). ResultsSequence quality processing and analysisData from the sequencing of the hypervariable region V4 of the bacterial 16S rRNA gene generated 436,935 crude sequences apart from the 6 Capra hircus samples, which had an average of 72,822.5 sequences per sample (ranging from 66,337 to 82,616). After data reduction by quality filtering using QIIME v 1.9.1., 157,950 high-quality sequences were generated (representing ~49% of the total sequences), with an average read length of 250 bp, scores quality (Q ≥ 25), length ≥ 150 bp, and chimera detection, with an average of 26,325 sequences per sample (ranging from 20,998 to 32,803; Table 2). Table 1. Feeding programming based on cactus, Salicornia, and native grass.
Table 2. Total number of sequences with low- and high-quality bacteria associated with IonTorrent PGM sequencing and 97% identification.
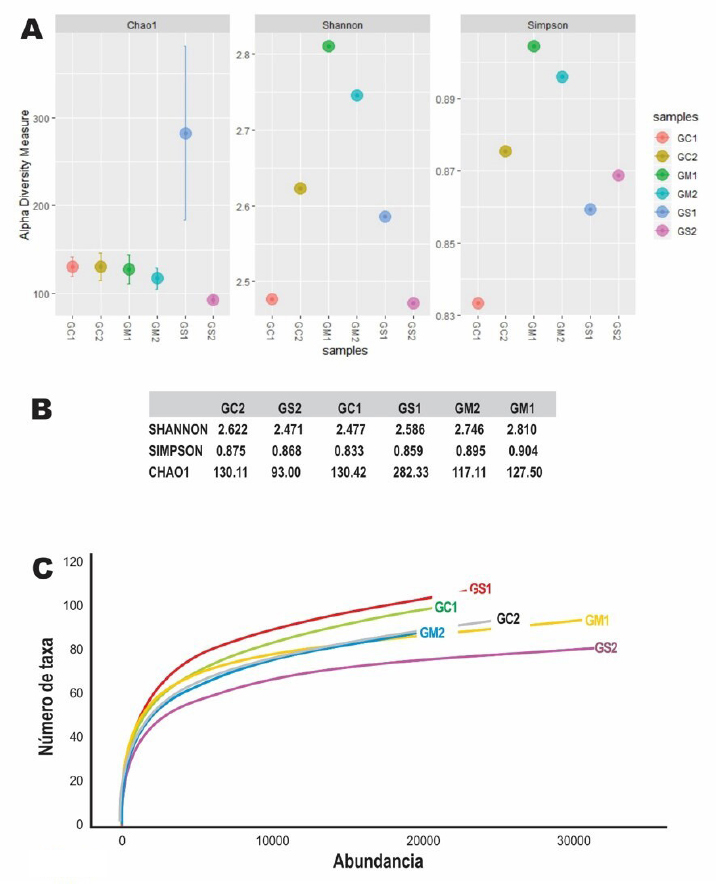
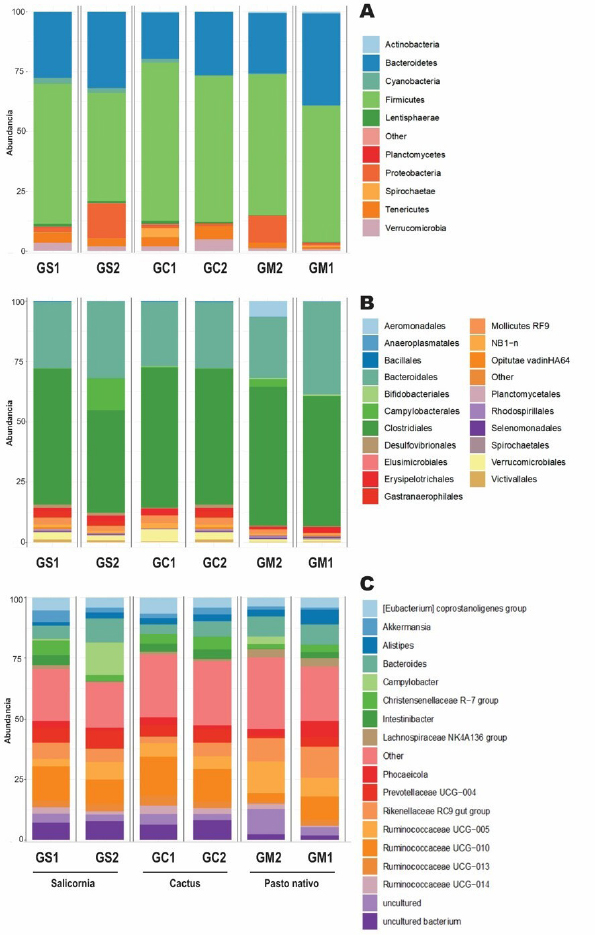
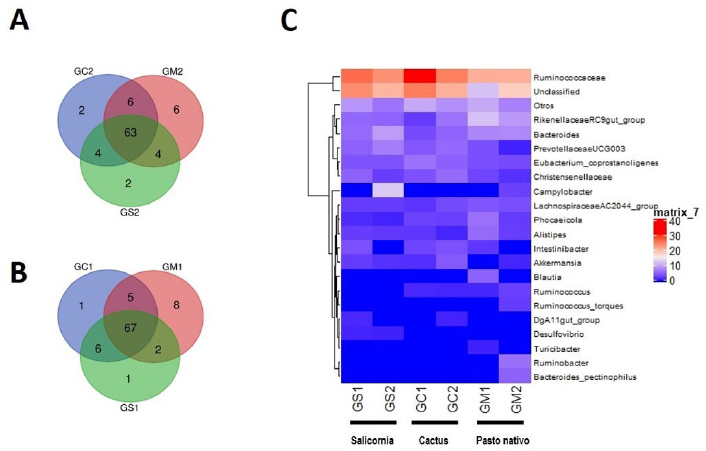
Analysis of the bacterial communityThe Shannon and Simpson diversity indices were slightly higher in GM1 and GM2 relative to the type of native grass compared to the other diets. With respect to the cactus diet, the bacterial diversity increased slightly with age; On the other hand, there was no significant difference with the Salicornia diet. The species richness observed and estimated (Chao1), compared between the six treatments, that GS1 had a significant variation compared to the other samples that did not have differentiations. Regarding its abundance, GS2 and GM1 related to the diet with Salicornia and native grass, respectively, showed a higher proportion than the other treatments; however, they decreased when related to the number of OTUs where GS1 showed greater bacterial diversity (Fig. 1). Taxonomic classificationThe taxonomic classification was distributed from the relative taxonomic abundances. A total of 10 phyla, 30 orders, and 95 genera greater than 1% of the total sequences were revealed. The 10 most abundant phyla were Acidobacteria, Bacteroidetes, Cyanobacteria, Lentisphaerae, Planctomycetes, Firmicutes, Proteobacteria, Spirochaetes, Tenericutes, and Verrucomicrobia. Among groups GC2, GS2, GC1, GS1, GM2, and GM1, Bacteroidetes (abundance of 28.8%, 33.1%, 21.1%, 29.9%, 26.1%, and 40.1%, respectively), Firmicutes (58.5%, 42.7%, 62.5%, 55.3%, 57.7%, and 55.2%, respectively), Proteobacteria (1.0%, 15.4%, 1.2%, 2.2%, 11.6%, and 0.9%, respectively), and Verrucomicrobia (5.4%, 2.1%, 2.3%, 3.6%, 1.3%, and 0.8%, respectively) were the most abundant, while Lentisphaerae, Spirochaetes, and Tenericutes were the least representative. A total of 30 OTUs at the order level were detected in the taxonomic analysis among groups GC2, GS2, GC1, GS1, GM2 and GM1; Bacteroidales (abundance of 28.8%, 33.3%, 21.1%, 29.9%, 26.1%, and 40.1%, respectively), Clostridiales (55.4%, 39.9%, 60.8%, 53.7%, 56.6%, and 52.5%, respectively), Verrucomicrobiales (5.3%, 2.0%, 2.2%, 3.1%, 1.3%, and 0.8%, respectively), and Campylobacterales (7.5%, 0.6%, 14.2%, 0.0%, 0.1%, 3.3%, and 0.1%, respectively) were the most abundant. While in groups GM2, GS2, GC1, and GM1, the taxa Methanobacteriales, Methanomicrobiales, Bifidobacteriales, Bacteriales, Gastranaerophilales, Elusimicrobiales, Bacillales, Clostridiales, Erysipelotrichales, Selenomanadales, Vicivallales, Planctomycetales, Rhodospirillales, Anaeroplasmatales, and Mollicutes were the most abundant. A total of 95 OTUs at the genus level were detected in the taxonomic analysis; in groups GC2, GS2, GC1, GS1, GM2, and GM1, Ruminococcaceae UGC-010, Campylobacter, Akkermansia, Coprostanoligenes group, Ruminococcaceae UGC-013, Ruminococcaceae UGC-005, Instestinibacter, Christensenellaceae R-7 group were the most abundant. It was shown that the greatest diversities of bacteria are found in GS2, GS1, GC1, and GC2; on the contrary, GM1 and GM2 presented less diversity of bacteria shared with the other groups Characterization of the core and unique microbiomeThe previously identified genera (Fig. 2) were studied to identify the microorganisms that are part of the core microbiome and the only one present in creole goats under different diets; it is worth mentioning that the core microbiome is considered if there is greater than or equal to 5 OTUs present in two or more samples during taxonomic analysis. For this study, the genera (Fig. 3) belonging to the core microbiome were identified in two groups: a) goats fed with cactus, Salicornia, and native grass; and b) kids fed with cactus, Salicornia, and native grass: In the first group of adult goats fed with cactus, Salicornia, and native grass, 63 genera belonging to the nucleus microbiome were characterized, among them the most abundant were Family XIII UGC-001, Candidatus, Soleaferrea, Ruminococcus 1, dgA-11 gut group, Dorea, Peptococcus, Phascolarctobacterium, Anaeroplasma, Ruminococcus 2, Prevotellaceae UGC-004, Aeriscardovia, Ruminiclostridium 9, Akkermansia, Phocaeicola, Turicibacter, Lachnospiraceae AC2044 group, Lachnospiraceae UGC-001, Lachnoclostridium, Ruminococcaceae UCG-010, uncultured bacterium, Lachnospiraceae FCS020 group, and Ruminiclostridium (Fig. 3). Meanwhile, in the diversity of the second group of kids fed with cactus, Salicornia, and native grass, 67 genera belonging to the core microbiome were characterized. Among them the most abundant were Bifidobacterium, Family XIII UGC-001, Candidatus, Soleaferrea, Ruminococcus 1, dgA-11 gut group, Peptococcus, Phascolarctobacterium, Victivallis, Terrisporobacter, Treponema 2, Anaeroplasma and Ruminococcus 2 (Fig. 3).
Fig. 1. Diversity indices between diets. A) Shannon, Simpson, and Chao1 diversity indices between samples using the Phyloseq package from R. B) Table of diversity indices between samples. C) Rarefaction curve showing bacterial diversity versus abundance number showing that the GS1 sample related to young goats using the vegan package using R.
Fig. 2. The relative abundance of bacterial communities on the effect of the Salicornia GS1 and GS2, cactus GC1 and GC2 diets compared to the native grass GM1 and GM2 diet in C. hircus. (A) Phylum level: all remaining taxa with abundance <1% are summarized as others. (B) Order level: all remaining taxa with abundance <1% are summarized as others. (C) Genus level: all remaining taxa with abundance <1% are summarized as others.
Fig. 3. Diagram illustrating the number of taxa in the superposition of the core bacterial communities. A) Goats fed with cactus, Salicornia, and native grass. B) Kids fed with cactus, Salicornia, and native grass. C) Heat map showing the gradients of higher and lower heat of bacteria between different diets. The previously identified core microbiome genera identified in both samples belonged to the order Alphaproteobacteria and Gammaproteobacteria. Then, we proceeded to characterize the unique microbiome of the first group of goats fed with cactus and native grass where GC1, GC2, GM1, and GM2 were 1, 1, 0, and 10 OTUs, respectively. The second group of goats fed with Salicornia and native grass was GM1, GM2, GS1, and GS2 with 1, 1, 0, and 0 OTUs, respectively. Afterward, the third group of goats fed with cactus and Salicornia was GC1, GC2, GS1, and GS2 with 0, 1, 2, and 0 OTUs, respectively (Fig. 3). The fourth group of goats fed with cactus and Salicornia was GC1, GM1, and GS1 were 1, 8, and 1 OTUs, respectively. Finally, the fifth group of goats fed with cactus and Salicornia was GC2, GM2, and GS2 was 2, 6, and 2 OTUs, respectively. It is worth mentioning that the bacterial genera identified in the nucleus microbiome of the first, second, third, and fourth group presented bacterial diversity, contrary to the fifth group where bacterial diversity was broadly demonstrated during the taxonomic analysis. DiscussionThe purpose of our research was to identify, characterize, and evaluate the microbiome of creole goats fed with different diets (cactus and Salicornia) compared to native grass. To do this, we resorted to the latest generation next-generation sequencing (NGS), a massive sequencing technique, which allowed us to characterize the bacterial microbiome at different taxonomic levels from phylum to genus. In the results, we find that Bacteroidetes and Firmicutes at the phylum level are the two most abundant OTUs in the microbiome of creole goats, ranging from 18% to 36% and from 47% to 66% respectively, influenced by the different diets between the samples and corroborated by previous research based on NGS technologies (Chaucheyras-Durand et al., 2016; Deusch et al., 2017; Cui et al., 2019). The intestinal tract of most ruminants is dominated by these types of phyla (Wang et al., 2019), and they play essential roles in carbohydrate, protein, and fiber metabolism (Huo et al., 2014). The percentage of Bacteroidetes, related to organic decomposition, decreases with increasing age, otherwise with Firmicutes, which are related to high fiber decomposition. Thus, we identified that those creole goats fed with cacti presented a greater abundance of Firmicutes concerning the other groups investigated as they contain a higher concentration of plant fibers. In this way, the host microbiome changes under the influence of high fiber diets, as many previous studies have been carried out in herbivores (Hook et al., 2011). The genus-level shows a significant change in the taxa altered by changing diets. Our results highlight variants of the genus Ruminococcaceae, related to cellulose degradation, as the most dominant in all samples, followed by Christensenellaceae, Rikenellaceae, and Prevotellaceae. But Campylobacter remarkably predominates in adult goats fed Salicornia. However, our results differ from previous research showing Prevotella as the most abundant genus (Pitta et al., 2010; Deusch et al., 2017; Wang et al., 2019; Ciu et al., 2019). On the other hand, Belzer and De Vos (2012) and Lagier et al. (2015) mention that Akkermansia is widely distributed in the intestinal tract of humans and animals and related to mucosal health (Derrien et al., 2016) and energy metabolism and inflammation markers (Schneeberger et al., 2015; Guo et al., 2017). Furthermore, studies report increased levels of pro-inflammatory TNF-α and IFN-γ are associated with higher amounts of Akkermansia sp. in breast milk (Collado et al., 2012). In our research, Akkermansia was identified with a higher presence in kids fed Salicornia. It is necessary to understand that the study of the bacterial microbiome allows us to know the diversity and richness of species related to the organism’s activity and behavior in the face of environmental conditions; in addition, a minimal change in the lifestyle of organisms could drastically change the bacterial microbiome. In conclusion, despite the reduced number of samples in the present study, we were able to identify that Firmicutes and Bacteroides play essential roles in the fiber-based feeding of creole goats, where a high population of both phyla would improve the production characteristics in creole goats such as weight gain, a proliferation of offspring, increase in milk, and meat production. AcknowledgmentsThis work was financed by contract No. 030-2016 from the National Institute of Agrarian Innovation (INIA), National Program of Agrarian Innovation (PNIA), UPMSI/IE, World Bank, and Inter-American Development Bank. The authors thank Rosita Morocho, Ivan Alama, Raúl Aponte, and Reyner Seminario for their support in the handling and feeding of the goats. Conflict of interestThe authors declare that there is no conflict of interest. ReferencesAllali, I., Arnold, J.W., Roach, J., Cadenas, M.B., Butz, N., Hassan, H.M. and Azcarate-Peril, M.A. 2017. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 17(1), 194. Arroyo, B.O. 1998. Producción de caprinos. Ed., Procabra. Lima, Perú, pp: 33. Benson, A.K., Kelly, S.A., Legge, R., Ma, F., Low, S.J., Kim. J. and Kachman, S.D. 2010. Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors. Proc. Natl. Acad. Sci. U.S.A. 107(44), 18933–18938. Belzer, C. and De Vos, W.M. 2012. Microbes inside from diversity to function: the case of Akkermansia. ISME J. 6, 1449–1458. Bokulich, N.A., Subramanian, S., Faith, J.J., Gevers, D., Gordon, J.I., Knight, R. and Caporaso JG. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10(1), 57–59. Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg-Lyons, D., Lozupone, C.A., Turnbaugh, P.J. and Knight, R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108(1), 4516–4522. Chaucheyras-Durand, F., Ameilbonne, A., Bichat, A., Mosoni, P., Ossa, F. and Forano, E. 2016. Live yeasts enhance fibre degradation in the cow rumen through an increase in plant substrate colonization by fibrolytic bacteria and fungi. J. Appl. Microbiol. 120(3), 560–570. Collado, M.C., Laitinen, K., Salminen, S. and Isolauri, E. 2012. Maternal weight and excessive weight gain during pregnancy modify the immunomodulatory potential of breast milk. Pediatr. Res. 72, 77–85. Cui, X., Wang, Z., Yan, T., Chang, S., Wang, H. and Hou, F. 2019. Rumen bacterial diversity of Tibetan sheep (Ovis aries) associated with different forage types on the Qinghai-Tibetan Plateau. Can. J. Microbiol. 65(12), 859–869. Derrien, M., Belzer, C. and De Vos, W.M. 2016. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog; doi:10.1016/j.micpath.2016.02.005. Deusch, S., Camarinha-Silva, A., Conrad, J., Beifuss, U., Rodehutscord, M. and Seifert, J. 2017. A structural and functional elucidation of the rumen microbiome influenced by various diets and microenvironments. Front. Microbiol. 8, 1605. Edgar, R.C., Haas, B.J., Clemente, J.C., Quince, C. and Knight, R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27(16), 2194–2200. Egan, A.R. 2005. Experimental designs for rumen microbiology. In Methods in gut microbial ecology for ruminants. Dordrecht, Netherlands: Springer, pp: 3–19. Frese, S.A., Parker, K., Calvert, C.C. and Mills, D.A. 2015. Diet shapes the gut microbiome of pigs during nursing and weaning. Microbiome 3(1), 28. Funkhouser, L.J. and Bordenstein, S.R. 2013. Mom knows best: the universality of maternal microbial transmission. PLoS Biol. 11(8):e1001631. Guevara, J.C. and Estevez, O.R. 2001. Opuntia spp. for fodder and forage production in Argentina: experiences and prospects. FAO Plant Production and Protection Papers, Mendoza, Argentina, pp: 63–72. Guo, X.F., Li, S.H., Zhang, J.C., Wu, F.F., Li, X.C., Wu, D. and Peng, Y.Z. 2017. Genome sequencing of 39 Akkermansia muciniphila isolates reveals its population structure, genomic and functional diversity, and global distribution in mammalian gut microbiota. BMC Genomics 18, 800; doi:10.1186/s12864-017-4195-3 Hook, S.E., Steele, M.A., Northwood, K.S., Dijkstra, J., France, J., Wright, A.D.G. and McBride, B.W. 2011. Impact of subacute ruminal acidosis (SARA) adaptation and recovery on the density and diversity of bacteria in the rumen of dairy cows. FEMS Microbiol. Ecol. 78(2), 275–284. Huang, K.W., Liu, T.C., Liang, R.Y., Chu, L.Y., Cheng, H.L., Chu, J.W. and Hsiao, Y.Y. 2018. Structural basis for overhang excision and terminal unwinding of DNA duplexes by TREX1. PLoS Biol. 16(5), e2005653. Huo, W., Zhu, W. and Mao, S. 2014. Impact of subacute ruminal acidosis on the diversity of liquid and solid-associated bacteria in the rumen of goats. World J. Microbiol. Biotechnol. 30(2), 669–680. Jiang, S., Huo, D., You, Z., Peng, Q., Ma, C., Chang, H. and Zhang, J. 2020. The distal intestinal microbiome of hybrids of Hainan black goats and Saanen goats. PLoS One 15(1), e0228496. Lagier, J.C., Hugon, P., Khelaifia, S., Fournier, P.E., La Scola, B. and Raoult, D. 2015. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin. Microbiol. Rev. 28, 237–264. McFall-Ngai, M., Hadfield, M.G., Bosch, T.C., Carey, H.V., Domazet-Lošo, T., Douglas, A.E. and Hentschel, U. 2013. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. U.S.A. 110(9), 3229–3236. McSweeney, C.S., Denman, S.E. and Mackie, R.I. 2005. Rumen bacteria. In Methods in gut microbial ecology for ruminants. Dordrecht, Netherlands: Springer, pp: 23–37. Pitta, D.W., Pinchak, E., Dowd, S.E., Osterstock, J., Gontcharova, V., Youn, E., Dorton, K., Yoon, I., Min, B.R., Fulford, J.D., Wickersham, T.A. and Malinowski, D.P. 2010. Rumen bacterial diversity dynamics associated with changing from bermudagrass hay to grazed winter wheat diets. Microb. Ecol. 59, 511–522. Schneeberger, M., Everard, A., Gómez-Valadés, A.G., Matamoros, S., Ramírez, S., Delzenne, N.M. and Cani, P.D. 2015. Akkermansia muciniphila inversely correlates with the onset of inflammation, altered adipose tissue metabolism and metabolic disorders during obesity in mice. Sci. Rep. 5, 16643; doi:10.1038/srep16643 Wang, L., Liu, K., Wang, Z., Xue, B., Peng, Q. and Jin, L. 2019. Bacterial community diversity associated with different utilization efficiencies of nitrogen in the gastrointestinal tract of goats. Front. Microbiol. 10, 239. | ||
| How to Cite this Article |
| Pubmed Style Domínguez FF, Crisanto MEV, Castro RLS, Rojas LV, Cuba VMB, Santos GRS, Salazar MWS, . Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets. Open Vet J. 2022; 12(1): 61-68. doi:10.5455/OVJ.2022.v12.i1.7 Web Style Domínguez FF, Crisanto MEV, Castro RLS, Rojas LV, Cuba VMB, Santos GRS, Salazar MWS, . Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets. https://www.openveterinaryjournal.com/?mno=127384 [Access: July 01, 2025]. doi:10.5455/OVJ.2022.v12.i1.7 AMA (American Medical Association) Style Domínguez FF, Crisanto MEV, Castro RLS, Rojas LV, Cuba VMB, Santos GRS, Salazar MWS, . Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets. Open Vet J. 2022; 12(1): 61-68. doi:10.5455/OVJ.2022.v12.i1.7 Vancouver/ICMJE Style Domínguez FF, Crisanto MEV, Castro RLS, Rojas LV, Cuba VMB, Santos GRS, Salazar MWS, . Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets. Open Vet J. (2022), [cited July 01, 2025]; 12(1): 61-68. doi:10.5455/OVJ.2022.v12.i1.7 Harvard Style Domínguez, F. F., Crisanto, . M. E. V., Castro, . R. L. S., Rojas, . L. V., Cuba, . V. M. B., Santos, . G. R. S., Salazar, . M. W. S. & (2022) Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets. Open Vet J, 12 (1), 61-68. doi:10.5455/OVJ.2022.v12.i1.7 Turabian Style Domínguez, Fredy Fabián, Milly Edith Vega Crisanto, Rosa Liliana Solís Castro, Lourdes Vásquez Rojas, Vanessa Miluska Baylon Cuba, Gabriela Raquel Sucapuca Santos, Marcos Walter Sanjinez Salazar, and Carlos Alberto Luque Ramos And Eric Mialhe. 2022. Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets. Open Veterinary Journal, 12 (1), 61-68. doi:10.5455/OVJ.2022.v12.i1.7 Chicago Style Domínguez, Fredy Fabián, Milly Edith Vega Crisanto, Rosa Liliana Solís Castro, Lourdes Vásquez Rojas, Vanessa Miluska Baylon Cuba, Gabriela Raquel Sucapuca Santos, Marcos Walter Sanjinez Salazar, and Carlos Alberto Luque Ramos And Eric Mialhe. "Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets." Open Veterinary Journal 12 (2022), 61-68. doi:10.5455/OVJ.2022.v12.i1.7 MLA (The Modern Language Association) Style Domínguez, Fredy Fabián, Milly Edith Vega Crisanto, Rosa Liliana Solís Castro, Lourdes Vásquez Rojas, Vanessa Miluska Baylon Cuba, Gabriela Raquel Sucapuca Santos, Marcos Walter Sanjinez Salazar, and Carlos Alberto Luque Ramos And Eric Mialhe. "Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets." Open Veterinary Journal 12.1 (2022), 61-68. Print. doi:10.5455/OVJ.2022.v12.i1.7 APA (American Psychological Association) Style Domínguez, F. F., Crisanto, . M. E. V., Castro, . R. L. S., Rojas, . L. V., Cuba, . V. M. B., Santos, . G. R. S., Salazar, . M. W. S. & (2022) Metagenomic analysis of the intestinal microbiome in goats on cactus and salicornia based diets. Open Veterinary Journal, 12 (1), 61-68. doi:10.5455/OVJ.2022.v12.i1.7 |

