




| Original Article | ||
Open Vet J. 2022; 12(6): 797-805 Open Veterinary Journal, (2022), Vol. 12(6): 797–805 Original Research Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy maresPamela Thomson1, Josefina Pareja1, Andrea Núñez2,3, Rodrigo Santibáñez4 and Rodrigo Castro2*1Laboratorio de Microbiología Clínica y Microbioma, Escuela de Medicina Veterinaria, Facultad de Ciencias de la Vida, Universidad Andrés Bello, Santiago, Chile 2Escuela de Medicina Veterinaria, Facultad de Recursos Naturales y Medicina Veterinaria, Universidad Santo Tomás, Santiago, Chile 3Facultad de Medicina Veterinaria y Agronomía, Universidad de las Américas, Santiago, Chile 4Department of Chemical and Bioprocess Engineering, Escuela de Ingeniería, Pontificia Universidad Católica de Chile, Santiago, Chile Submitted: 26/04/2022 Accepted: 10/10/2022 Published: 07/11/2022 *Corresponding Author: Rodrigo Castro. Escuela de Medicina Veterinaria, Facultad de Recursos Naturales y Medicina Veterinaria, Universidad Santo Tomás, Santiago, Chile. Email: rodrigocastro [at] santotomas.cl © 2022 Open Veterinary Journal
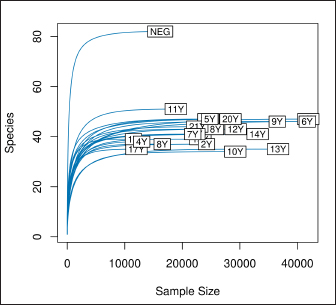
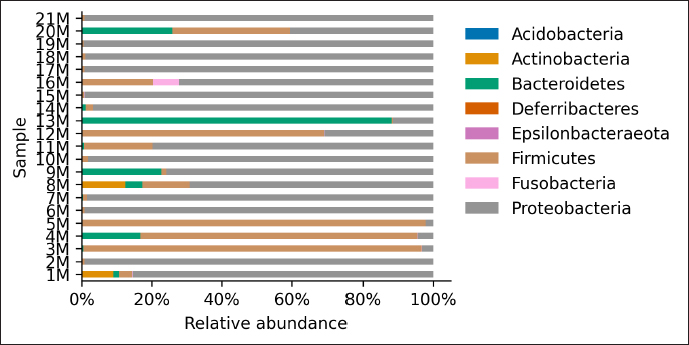
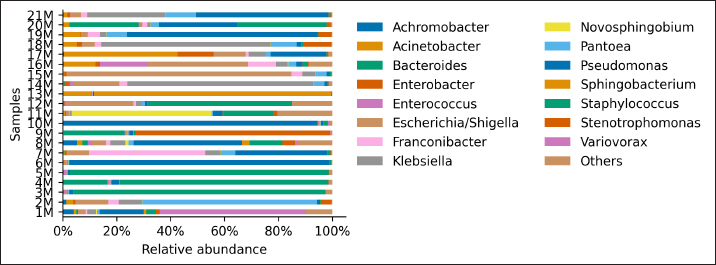
AbstractBackground: Culture-independent techniques have made it possible to expand the knowledge about the composition of bacterial communities present in the healthy uterus and their role in health and disease, mainly in humans. However, in animals like mares, there is a dearth of information regarding this area. Aim: To narrow this knowledge gap, the objective of this study was to identify and characterize the composition and function of the uterine microbiome of a group of Chilean purebred mares (CPM), an equine breed with the oldest genealogical record in South America and an economical important reproductive industry. Methods: From uterine biopsy samples obtained during estrus, DNA extraction and targeted sequencing were performed to investigate the bacterial diversity and its probable metabolic function. Results: CPM biopsy samples were characterized by having a varied microbial composition, where the four most relatively abundant phyla were Proteobacteria (69.6%), Firmicutes (21.1%), Bacteroidetes (7.8%), and Actinobacteria (1.06%); which made up 99.6% of the total identified phyla. In contrast, Actinobacteria and Fusobacteria were the phyla not identified in all samples. Of a total of 59 genera identified across all samples, Staphylococcus was the most abundant genus with an average relative abundance of 18.88%, followed by Pseudomonas (17.9%), Escherichia/Shigella (10.42%), and Klebsiella (9.92%). Conclusion: These findings contribute to the knowledge of microbes’ presence in the uterus, while future studies are required to demonstrate the role of these microorganisms in health and disease. Keywords: Uterine microbes, Mare, Metabolic pathways. IntroductionDuring the last years, it has been shown that the healthy uterine epithelium is not sterile as it was believed during the 1990s (Tessier, 1900; Baker et al., 2018). The microbes identified from uterine samples are taxonomically diverse and some of them are capable of growing in culture (Costa and Weese, 2019; Shanahan et al., 2021). However, culture-dependent microbial characterization is limited since not all microorganisms manage to grow in vitro (Shanahan et al., 2021). Difficult-to-grow microorganisms have redirected the study of the microbiome to use 16S ribosomal RNA sequencing (Peterson et al., 2009), which has made it possible to detect bacteria that, by their nature, take a long time or do not grow in conventional cultures (Ferris et al., 2010; Yang et al., 2017). In humans, evidence of the presence of endometrial microbes suggests they might play a relevant role at the time of embryo implantation, through the regulation of endometrial cell function and the local immunity response. It might help also to prevent the growth of pathogenic microorganisms through the protection of the epithelium (Benner et al., 2018). However, the complete role of microbiomes in the reproductive tract is still not fully understood. It has been suggested that a change in microbial composition and the presence or absence of some bacterial species could help in the maintenance and completion of a pregnancy (Heil et al., 2019; Lozano et al., 2021). In mares, a correlation has been observed between microbes in the endometrium and the reproductive state, where the presence of bacteria of the phyla Proteobacteria and Bacteroidetes was positively associated with pregnancy health (Sathe et al., 2017). In addition, other authors have identified from uterine samples of healthy mares the presence of bacteria of the phyla Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria (Sathe et al., 2017; Schnobrich et al., 2017; Heil et al., 2019). Therefore, the identification and characterization of microbes from the uterine epithelium can help to understand its role in health and disease. However, the presence of microbes from uterine samples does not relate to an established microbiome, and the identified taxa might come from contaminated reagents and environments other than the uterus (Kim et al., 2017; Olomu et al., 2020; Blaser et al., 2021). Several studies have directly or indirectly associated dysbiosis or bacterial abundance alterations with subfertility (Heil et al., 2019), premature birth (Puente et al., 2020; Morimune et al., 2021), and early mortality in different species, including humans (Goldenberg et al., 2000; Lawn et al., 2005; Moore et al., 2017; Yang et al., 2017; Lyman et al., 2019; Bardos et al., 2020). In mares, bacterial endometritis is recognized as the main cause of infertility, causing a decrease in reproductive efficiency and therefore, a substantial economic loss (Causey, 2006; Troedsson and Woodward, 2016; Ferris et al., 2017; Heil et al., 2019; Gallego et al., 2020). Culture-dependent studies of the uterine epithelium have indicated a predominance of Enterobacteriaceae in mares with endometritis (Ferrer and Palomares, 2018) a family of the gamma-proteobacteria phylum that include Escherichia coli, Salmonella, Shigella, and Klebsiella. In Chilean purebred horses, it is common for them to start their reproductive life late, causing elderly and multiparous mares to remain in use as breeders. Therefore, a positive association between endometritis, age, and the number of deliveries has been reported, possibly related to E. coli being the most frequently isolated microorganism using conventional culture techniques (Morales and Castro, 2018). Despite the economic importance of Chilean purebred mares (CPM) in Chile, there are no previous reports describing the identification of microbes from uterine samples. In this study, we identified and predicted the metabolic capabilities of bacteria from biopsy samples for a group of CPM, using targeted next-generation sequencing and bioinformatics analyses. Materials and MethodsThis study was carried out in Chile, in the Maule region (35°25ʹS, 71°39ʹW) during the month of October 2021, which presented days of 14.1 hours of light and 9.9 hours of darkness, with an average temperature of 14°C (57.2 F). Inclusion criteria were clinically healthy mares in the ovulatory phase and without antibiotic treatment at least 3 months before sampling. The sampled group consisted of 21 mares between 4 and 23 years old (10.76 ± 6.6 years old), registered in the genealogical records of the National Society of Agriculture of Chile (https://www.sna.cl). The mares were kept for 1 month in the sportive break and fed in a mixed meadow of ryegrass and white clover, with free access to water. In the reproductive records, only one mare had not started her reproductive life while all the other mares had already foaled one or more times. No mare presented records of abortions, embryonic losses, endometritis, dystocia, or any reproductive pathology. The mares underwent a gynecological examination to determine the reproductive phase employing a transrectal ultrasound (Chison Eco 6 ultrasound, 5 MHz linear transducer). To avoid contamination as much as possible, the tail was covered with sterile gauze and the perineum and vulva were washed with soap and water until clean. When sampling, the operator was asked to wear sterile rectal examination gloves (Ferrer and Palomares, 2018). A uterine sample was obtained with a double protection sterile swab. The sample was stained with panoptic staining (Diff Quick) for cytology evaluation under light microscopy, considering that the healthy endometrium presents <2% polymorphonuclear cells of the total cells observed (Morales and Castro, 2018). Healthy mares underwent a sterile forceps uterine biopsy through a sterile vaginoscope (Kruuse Catalogue No. 141965) (Ferris, 2016). Each sample was introduced into conical bottom tubes with RNA later (Sigma Aldrich) and immediately transferred to be processed in the clinical microbiology and microbiome laboratory of the Universidad Andrés Bello, Chile. Total DNA extraction was performed with a commercial extraction kit (Quick-DNA Microprep Plus Kit, Zymo Research, Irvine, CA) following the manufacturer’s instructions. Before the extraction, each sample was crushed and then vigorously shaken using a Genie disruptor device (Scientific Industries, Bohemia, NY) (Medina et al., 2017). The DNA obtained from each sample was diluted to 20 ng/µl in nuclease-free water (NanoDrop 2000c; Thermo Fisher Scientific, Wilmington, DE), at this stage, negative and positive controls were included and underwent targeted amplification and sequencing performed by Molecular Research LP (MR-DNA, Shallowater, TX). The variable V3–V4 region of the 16S rRNA gene was amplified using the 341 F and 785 R primers (Comeau et al., 2017). A polymerase chain reaction (PCR) reaction was run for 30 cycles using the HotStarTaq Plus Master Mix Kit (Qiagen, Germantown, MD). After amplification, the PCR products were verified on a 2% agarose gel. PCR products were pooled and purified using calibrated Ampure XP microspheres (Agencourt Bioscience Corporation, Beverly, MA). The combined and purified PCR products were used to prepare a DNA library using the TruSeq DNA LT Sample Preparation Kit (Illumina, San Diego, CA) following the manufacturer’s instructions. Sequencing was performed using the MiSeq platform (Illumina, San Diego, CA). The raw DNA sequences provided by the external service were analyzed using the open-source bioinformatics tool QIIME version 1.8.0 (Caporaso et al., 2010), DADA2, and PICRUSt2 (Callahan et al., 2016). Each sample was demultiplexed into individual files and barcodes were removed from the 5ʹend of each read (via the demultiplex_fasta.py script). The demultiplexed sequences were uploaded to the European Nucleotide Archive under the project code PRJEB47718. All reads were processed using the DADA2 v1.10 R package (Callahan et al., 2016), following a modified procedure. Briefly, the sequences were quality filtered to remove reads with indeterminate base calls and trimmed down to 220 nucleotides. Then, all filtered reads were used to estimate a sequencing error model. The model was used to infer Amplicon Sequence Variants (ASV) (Callahan et al., 2017) for each unique read per sample. Each unique ASV per sample was assigned to a bacterial taxonomy employing a Naïve Bayesian classifier (Wang et al., 2007) and the SILVA database version 132 (Quast et al., 2013; Yilmaz et al., 2014). Bioinformatics processing of a negative control composed of eluted nuclease-free water through the extraction kit identified Neorhizobium, Pseudarthrobacter, Streptococcus, and unclassified genera as possible contaminants contributing each more than 1% of the total reads in the negative control. Therefore, assigned reads for each biopsy sample to Neorhizobium, Pseudarthrobacter, Streptococcus, and unclassified genera were removed. Finally, PICRUSt2 (Douglas et al., 2020) was used to infer gene and metabolic pathways abundance considering each ASV and its abundance in a sample. Ethical approvalAll procedures performed using animals were revised and approved by the Scientific Committee of Ethics of the Central-South macrozone of Santo Tomás University, Chile. Authorization N° 60–21. ResultsAfter 16S rRNA sequencing, each uterine biopsy sample contained up to 40,000 reads and up to 50 ASVs per sample (Figure 1). The rarefaction curves showed saturation, indicating that the depth of sequencing was appropriate to describe the microbial composition in this group. Sequence analysis identified bacterial microorganisms from the Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, and Fusobacteria phyla. Acidobacteria and Deferribacteres were identified concurrently in only one sample. Proteobacteria was the most relatively abundant phylum with an average of 69.6%, followed by Firmicutes (21.1%) and Bacteroidetes (7.8%) (Figure 2). These three phyla make up 98.6% of the total identified bacteria and are present in all sampled mares. In contrast, the microorganism of the Actinobacteria and Fusobacteria were only identified in some mares. Overall, 59 different bacterial genera were identified considering all samples, with Staphylococcus being the most predominant genus, with an average relative abundance of 18.88%, followed by Pseudomonas (17.9%), Escherichia/Shigella (10.42%), and Klebsiella (9.92%) (Figure 3). These four dominant genera represented 57.12% of the total taxonomically assigned ASVs.
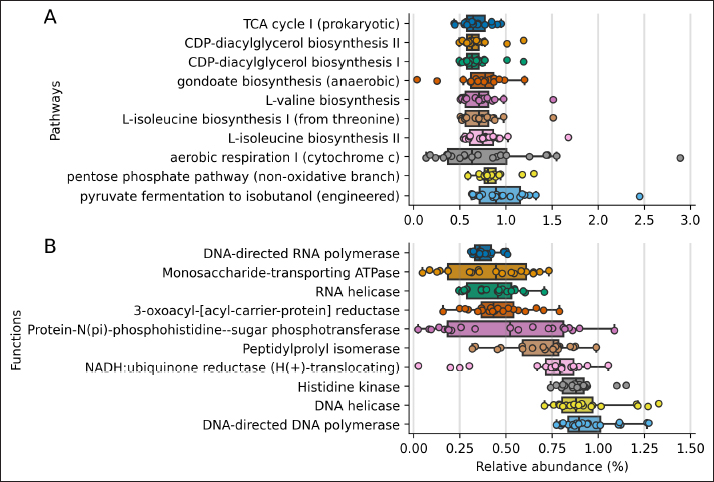
Fig. 1. Rarefaction curve. Number of identified ASVs as a function of the number of sequenced 16S amplicons. Finally, we inferred the abundance of genes associated with metabolic functions and pathways using PICRUSt2 for each sample (Figure 4). The 10 most abundant pathways contribute at least 0.6% on average to each sample, with fermentation and energy production, and amino acids and lipid biosynthesis among the inferred genomic contents. Regarding metabolic functions, the ten most abundant functions contribute at least 0.4% to each sample and are related to the DNA and RNA synthesis, transport of monosaccharides, and lipid biosynthesis. DiscussionIn this study, we report the taxonomy and abundance of bacterial groups present in the uterine epithelium of healthy mares and the inferred abundance of metabolic functions of such microbiomes. Results showed that the most abundant phyla were Proteobacteria, Firmicutes, and Bacteroidetes, and coincide with previous reports (Sathe et al., 2017; Heil et al., 2019). In addition, bacteria from the Proteobacteria phylum have been identified as dominant in the equine endometrium during the estrous phase (Heil et al., 2018), and the estrous cycle correlates with the diversity of identified bacteria from the uterus (Heil et al., 2019). Similar to our results, research carried out to evaluate the healthy endometrium of pandas and bitches indicates that the most abundant phyla are Proteobacteria, Actinobacteria, and Bacteroides (Yang et al., 2017; Lyman et al., 2019). On the contrary, Firmicutes has been described as the most predominant phylum in the endometrium of cows (Heil et al., 2019).
Fig. 2. Relative abundance of representative taxa identified from uterine biopsy samples at the phylum level. The figure shows the average proportion of Proteobacteria, Firmicutes, Actinobacteria, Bacteroidetes, Acidobacteria, Fusobacteria, and Deferribacteres.
Fig. 3. Relative abundance of representative taxa identified from uterine biopsy samples at the genus level. The figure shows the average proportion of 15 different bacterial genera that were detected over a 1% threshold considering all samples. The remaining 44 genera were grouped into “others.” Regarding genera abundance, Pseudomonas and Acinetobacter, respectively showed 17.9% and 4.18% average relative abundance across samples; previously these genera have also been described as part of the eyeball and gut microbiome of healthy horses (LaFrentz et al., 2020; Park et al., 2021; Santibáñez et al., 2022). These genera were represented by Acinetobacter genomospecies 3, Acinetobacter junii, Pseudomonas aeruginosa, and Pseudomonas amygdali; these species have been isolated from hospital environments (Horrevorts et al., 1995; Bassetti et al., 2021) and they have been associated with antibiotic resistance (Bello-López et al., 2020; Panzuti et al., 2020; Gruszecka et al., 2021). In horses, Acinetobacter baumannii and Acinetobacter calcoaceticus have been isolated from a venous catheter-associated with thrombophlebitis (Vaneechoutte et al., 2000). On the other hand, P. aeruginosa has been related to pyometra in mares and vasculitis in foals, among other reports (Köhne et al., 2020; Panzuti et al., 2020). The WHO warns about A. baumannii and P. aeruginosa as critical microorganisms in terms of their resistance profile (Allen et al., 2011; OMS, 2017), species belonging to the same genera found in this research. It is necessary to deepen the knowledge about these microorganisms in the equine and its clinical environment, to elucidate what is the role that they play in the uterine environment of mares and the potential transmission to humans and their environment (Malaluang et al., 2021). On the other hand, Staphylococcus equorum, Shigella sonnei, Arthrobacter ramosus, and P. amygdali were also species with a high relative abundance. All of them are identified for the first time, as a colonizer of the uterus of CPM. S. equorum is a species isolated from the skin of healthy horses (Jeong et al., 2017) and its most important role is related to the food industry (Leroy et al., 2009; Irlinger et al., 2012). On the other hand, S. sonnei can cause hemorrhagic diarrhea in immunosuppressed people (Torraca et al., 2020), and there is no evidence of the effects of Shigellosis in horses. Both A. ramosus and Pseudomonas amygdali are saprophytic species of soil and plants, without any role described as pathogens (Bafana et al., 2010; Chai et al., 2020; Jia et al., 2022). In mares suffering from bacterial endometritis, E. coli is the most frequently isolated bacterium, followed by Streptococcus equi subsp. zooepidemicus and Staphylococcus spp. (Gallego et al., 2020; Morris et al., 2020). Klebsiella pneumoniae and coagulase-negative Staphylococcus have also been isolated from mares suffering endometritis (Sathe et al., 2017; Morales and Castro, 2018; Omar et al., 2022). Other bacteria, fungi, and yeasts may also cause infectious endometritis, some of these other infections are iatrogenically introduced environmental contaminants, venereal pathogens, or secondary to over-treatment with antibiotics. These pathogens may be rare, but highly pathogenic. For example, P. aeruginosa can be either an opportunistic environmental or venereal pathogen. Other bacteria, such as streptococci, can remain dormant in the uterus and be activated by processes such as persistent endometritis. Therefore, in bacterial endometritis, it is not known whether the bacteria present in the uterus are there as the main cause of the problem or secondary to it, and it is essential to reduce risk factors before and during all stages of reproductive management (Morris et al., 2020).
Fig. 4. PICRUSt2 inferred abundance of metabolic genes per sample. Distribution of the 10 most abundant metabolic genes associated with pathways (A) and functions (B) using the MetaCyc annotation. The shown pathways contribute 0.67% and functions contribute 0.38%, on average to each sample. The predicted metabolic pathways present in the group of mares studied were those related to common bacterial metabolic functions, such as cell wall and membrane biosynthesis, carbon catabolism, and pyrimidine recovery (Mio et al., 1999; Rodionova et al., 2012; Cámara et al., 2013; Okesli et al., 2017). The unaltered abundance of pathways related to virulence factors is consistent with the fact that the group studied was made up of mares without uterine pathologies. In perspective, a study that compares the microbiome of a group of mares without reproductive pathologies with another that presents reproductive disorders would be necessary; this would help to understand the role of bacteria in the reproductive environment in states of health and disease; considering a strict sampling protocol, to avoid cross-contamination of the sample with fecal material (Kim et al., 2017; Olomu et al., 2020; Blaser et al., 2021). ConclusionThe targeted sequencing and bioinformatics analysis of 21 uterus biopsy samples of CPM identified seven phyla and 59 genera. Proteobacteria was the phylum with the highest relative abundance, while Staphylococcus was the genus most represented and identified in all samples. AcknowledgmentsThe authors acknowledge Agencia Nacional de Investigación y Desarrollo (ANID, Ministerio de Ciencia, Tecnología, Conocimiento e Innovación, Chile), Project PAI #77190079. Conflict of interestThe authors have no conflict of interest to declare. Author contributionsPamela Thomson: Project Administration, Conceptualization, Methodology, Validation, Formal Analysis, Data Curation, Writing – Original Draft, Review & Editing. Funding Acquisition, Investigation. Josefina Pareja: Data Curation, Writing – Original Draft. Andrea Núñez: Investigation, Writing – Original Draft, Data Curation, Methodology. Rodrigo Santibáñez: Bioinformatics Analyses, Statistic Analyses, Writing – Review & Editing. Rodrigo Castro: Investigation, Conceptualization, Methodology, Validation, Data Curation, Review & Editing. ReferencesAllen, J.L., Begg, A.P. and Browning, G.F. 2011. Outbreak of equine endometritis caused by a genotypically identical strain of Pseudomonas aeruginosa. J. Vet. Diagn. Invest. 23, 1236–1239. Bafana, A., Krishnamurthi, K., Patil, M. and Chakrabarti, T. 2010. Heavy metal resistance in Arthrobacter ramosus strain G2 isolated from mercuric salt-contaminated soil. J. Hazard. Mater. 177, 481–486. Baker, J.M., Chase, D.M. and Herbst-Kralovetz, M.M. 2018. Uterine microbiota: residents, tourists, or invaders? Front. Immunol. 9, 1–16. Bardos, J., Fiorentino, D., Longman, R.E. and Paidas, M. 2020. Immunological role of the maternal uterine microbiome in pregnancy: pregnancies pathologies and alterated microbiota. Front. Immunol. 10, 1–14. Bassetti, M., Echols, R., Matsunaga, Y., Ariyasu, M., Doi, Y., Ferrer, R., Lodise, T.P., Naas, T., Niki, Y., Paterson, D.L., Portsmouth, S., Torre-Cisneros, J., Toyoizumi, K., Wunderink, R.G. and Nagata, T.D. 2021. Efficacy and safety of cefiderocol or best available therapy for the treatment of serious infections caused by carbapenem-resistant gram-negative bacteria (CREDIBLE-CR): a randomised, open-label, multicentre, pathogen-focused, descriptive, phase 3 trial. Lancet. Infect. Dis. 21, 226–240. Bello-López, E., Rocha-Gracia, R.D.C., Castro-Jaimes, S., Cevallos, M.Á., Vargas-Cruz, M., Verdugo-Yocupicio, R., Sáenz, Y., Torres, C., Gutiérrez-Cázatrez, Z., Arenas-Hernández, M.M. and Lozano-Zarain, P. 2020. Antibiotic resistance mechanisms in Acinetobacter spp. strains isolated from patients in a paediatric hospital in Mexico. J. Glob. Antimicrob. Resist. 23, 120–129. Benner, M., Ferwerda, G., Joosten, I. and Van der Molen, R. 2018. Uterine microbiome and endometrial receptivity. How uterine microbiota might be responsible for a receptive, fertile endometrium. Hum. Reprod. Update. 24, 393–415. Blaser, M.J., Devkota, S., McCoy, K.D., Relman, D.A., Yassour, M. and Young, V.B. 2021. Lessons learned from the prenatal microbiome controversy. Microbiome 9, 1–7. Callahan, B.J., McMurdie, P.J., Rosen, M.J., Han, A.W., Johnson, A.J.A. and Holmes, S.P. 2016. DADA2: high-resolution sample inference from illumina amplicon data. Nat. Methods. 13, 581–583. Callahan, B.J., McMurdie, P.J. and Holmes, S.P. 2017. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME. J. 11, 2639–2643. Cámara, Y., González-Vioque, E., Scarpelli, M., Torres-Torronteras, J. and Martí, R. 2013. Feeding the deoxyribonucleoside salvage pathway to rescue mitochondrial DNA. Drug. Discov. Today. 18, 950–957. Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., Fierer, N., Gonzalez Pena, A., Goodrich, J.K., Gordon, J.I., Huttley, G.A., Kelley, S.T., Knights, D., Koenig, J.E., Ley, R.E., Lozupone, C.A., McDonald, D., Muegge, B.D., Pirrung, M., Reeder, J., Sevinsky, J.R., Turnbaugh, P.J., Walters, W.A., Widmann, J., Yatsunenko, T., Zaneveld, J. and Knight, R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 7, 335–336. Causey, R.C. 2006. Making sense of equine uterine infections: the many faces of physical clearance. Vet. J. 172, 405–421. Chai, A., Yuan, L., Li, L., Shi, Y., Xie, X., Wang, Q. and Li, B. 2020. Aerosol transmission of Pseudomonas amygdali pv. lachrymans in greenhouses. Sci. Total. Environ. 748, 141433. Comeau, A.M., Douglas, G.M. and Langille, M.G.I. 2017. Microbiome helper: a custom and streamlined workflow for microbiome research. MSystems 2, 1–11. Costa, M. and Weese, J.S. 2019. Methods and basic concepts for microbiota assessment. Vet. J. 249, 10–15. Douglas, G.M., Maffei, V.J., Zaneveld, J.R., Yurgal, S.N., Brown, J.R., Taylor, C.M., Huttenhower, C. and Langille, M.G.I. 2020. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. Ferrer, M. and Palomares R. 2018. Aerobic uterine isolates and antimicrobial susceptibility in mares with post-partum metritis. Equine. Vet. J. 50, 202–207. Ferris, R.A., Veir, J.K., Lappin, M.R. and Mccue, P.M. 2010. Development and clinical application of a broad range 16S quantitative PCR assay for detection of bacteria in the uterus of the mare. Anim. Reprod. Sci. 121, 98–100. Ferris, R.A. 2016. Endometritis: diagnostic tools for infectious endometritis. Vet. Clin. North. Am. Equine. Pract. 32, 481–498. Ferris, R.A., Palmer, B.A., Borlee, B.R. and McCue, P.M. 2017. Ability of chromogenic agar, MALDI-TOF, API 20E and 20 strep strips, and BBL crystal enteric and gram-positive identification kits to precisely identify common equine uterine pathogens. J. Equine. Vet. Sci. 57, 35–40. Gallego, R.S., Ruiz, A.F. and Ruiz, J.D. 2020. Frecuencia del aislamiento bacteriano y patrones de sensibilidad en yeguas criollas colombianas diagnosticadas con endometritis. Rev. Med. Vet. 1, 13–21. Goldenberg, R.L., Hauth, J.C. and Andrews, W.W. 2000. Intrauterine infection and preterm delivery. N. Engl. J. Med. 342, 1500–1507. Gruszecka, J., Filip, R. and Gutkowska, D. 2021. The state of microbiological cleanliness of surfaces and equipment of an endoscopic examination laboratory—data from a reference tertiary clinical endoscopy center in southern poland. Int. J. Environ. Res. Public. Health. 18, 6346. Heil, B.A., Thompson, S.K., Kearns, T.A., Davolli, G.M., King, G. and Sones, J.L. 2018. Metagenetic characterization of the resident equine uterine microbiome using multiple techniques. J. Equine. Vet. Sci. 66, 111. Heil, B.A., Paccamonti, D.L. and Sones, J.L. 2019. Role for the mammalian female reproductive tract microbiome in pregnancy outcomes. Physiol. Genomics. 51, 390–399. Horrevorts, A., Bergman, K., Kollee, L., Breuker, I., Tjernberg, I. and Dijkshoorn, L. 1995. Clinical and epidemiological investigations of Acinetobacter genomospecies 3 in a neonatal intensive care unit. J. Clin. Microbiol. 33, 1567–1572. Irlinger, F., Loux, V., Bento, P., Gibrat, J.F., Straub, C., Bonnarme, P., Landaud, S. and Monnet, C. 2012. Genome sequence of Staphylococcus equorum subsp. equorum Mu2, isolated from a French smear-ripened cheese. J. Bacteriol. 194, 5141–5142. Jeong, D.W., Heo, S., Ryu, S., Blom, J. and Lee, J.H. 2017. Genomic insights into the virulence and salt tolerance of Staphylococcus equorum. Sci. Rep. 7, 1–11. Jia, J., Copes, W., Phillips, K. and Lu, S. 2022. Complete genome sequence resource for Pseudomonas amygdali pv. loropetali strain AAC causing bacterial Gall of Loropetalum chinense. Plant. Dis. 106, 2502–2505. Kim, D., Hofstaedter, C.E., Zhao, C., Mattei, L., Tanes, C., Clarke, E., Lauder, A., Sherrill-Mix, S., Chehoud, C., Kelsen, J., Conrad, M., Collman, R.G., Baldassano, R., Bushman, F.D. and Bittinger, K. 2017. Optimizing methods and dodging pitfalls in microbiome research. Microbiome 5, 1–14. Köhne, M., Tönissen, A., Unruh, C., Pruß, D. and Sieme, H. 2020. Occurrence of intrauterine purulent concrements in a Maiden Mare—a case report. J. Equine. Vet. Sci. 95, 103278. LaFrentz, S., Abarca, E., Mohammed, H., Cuming, R. and Arias, C. 2020. Characterization of the normal equine conjunctival bacterial community using culture-independent methods. Vet. Ophthalmol. 23, 480–488. Lawn, J., Zupan, J. and Cousens, S. 2005. 4 million neonatal deaths: when? where? why? Lancet 365, 891–900. Leroy, S., Lebert, I., Chacornac, J.P., Chavant, P., Bernardi, T. and Talon, R. 2009. Genetic diversity and biofilm formation of Staphylococcus equorum isolated from naturally fermented sausages and their manufacturing environment. Int. J. Food. Microbiol. 134, 46–51. Lyman, C.C., Holyoak, G.R., Meinkoth, K., Wieneke, X., Chillemi, K.A. and DeSilva, U. 2019. Canine endometrial and vaginal microbiomes reveal distinct and complex ecosystems. PLoS One 14, 1–17. Lozano, F.M., Bernabeu, A., LLedo, B., Morales, R., Díaz, M., Aranda, F.I., Llacer, J. and Bernabeu, R. 2021. Characterization of the vaginal and endometrial microbiome in patients with chronic endometritis. Eur. J. Obs. Gynecol. Reprod. Biol. 263, 25–32. Malaluang, P., Wilén, E., Lindahl, J., Hansson, I. and Morrell, J. 2021. Antimicrobial resistance in equine reproduction. Animals 11, 1–13. Medina, D.A., Pedreros, J.P., Turiel, D., Quezada, N., Pimentel, F., Escalona, A. and Garrido, D. 2017. Distinct patterns in the gut microbiota after surgical or medical therapy in obese patients. PeerJ 5, e3443. Mio, T., Yamada-Okabe, T., Arisawa, M. and Yamada-Okabe, H. 1999. Saccharomyces cerevisiae GNA1, an essential gene encoding a novel acetyltransferase involved in UDP-N-acetylglucosamine synthesis. J. Biol. Chem. 274, 424–429. Moore, S.G., Ericsson, A.C., Poock, S.E., Melendez, P. and Lucy, M.C. 2017. Hot topic: 16S rRNA gene sequencing reveals the microbiome of the virgin and pregnant bovine uterus. J. Dairy. Sci. 100, 4953–4960. Morales, P.C. and Castro, R.A. 2018. Estimation of the uterine integrity in chilean purebred mares and its association with age and foaling number. Rev. Investig. Vet. Del. Perú. 29, 565–574. Morimune, A., Kimura, F., Nakamura, A., Kitazawa, J., Takashima, A., Amano, T., Kaku, S., Moritani, S., Kushima, R. and Murakami, T. 2021. The effects of chronic endometritis on the pregnancy outcomes. Am. J. Reprod. Immunol. 85, e13357. Morris, L., McCue, P. and Aurich, C. 2020. Equine endometritis: a review of challenges and new approaches. Reproduction 160, 95–110. Okesli, A., Khosla, C. and Bassik, M.C. 2017. Human pyrimidine nucleotide biosynthesis as a target for antiviral chemotherapy. Cur. Opi. Microbio. 48, 127–134. Olomu, I.N., Pena-Cortes, L.C., Long, R.A., Vyas, A., Krichevskiy, O., Luellwitz, R., Singh, P. and Mulks, M.H. 2020. Elimination of "kitome" and "splashome" contamination results in lack of detection of a unique placental microbiome. BMC. Microbiol. 20, 157. Omar, H., Hambidge, M., Firmanes, B., Shabandri, A. and Wilsher, S. 2022. Bacteria isolated from equine uteri in the united Arab emirates: a retrospective study. J. Equine. Vet. Sci. 115, 1–9. OMS. 2017. La OMS publica la lista de las bacterias para las que se necesitan urgentemente nuevos antibióticos. Comun. Prensa. OMS. Available via https://www.who.int/es/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (Accessed 17 July 2021). Panzuti, P., Ferrer, G.R., Mosca, M. and Pin, D. 2020. Equine pastern vasculitis in a horse associated with a multidrug-resistant Pseudomonas aeruginosa isolate. Vet. Dermatol. 31, 247–e55. Park, T., Cheong, H., Yoon, J., Kim, A., Yun, Y. and Unno, T. 2021. Comparison of the fecal microbiota of horses with intestinal disease and their healthy counterparts. Vet. Sci. 8, 113. Peterson, J., Garges, S., Giovanni, M., McInnes, P., Wang, L., Schloss, J.A., Bonazzi, V., McEwen, J.E., Wetterstrand, K.A., Deal, C., Baker, C.C., Di Francesco, V., Howcroft, TK., Karp, R.W., Lunsford, R.D., Wellington, C.R., Belachew, T., Wright, M., Giblin, C., David, H., Mills, M., Salomon, R., Mullins, C., Akolkar, B., Begg, L., Davis, C., Grandison, L., Humble, M., Khalsa, J., Little, A.R., Peavy, H., Pontzer, C., Portnoy, M., Sayre, M.H., Starke-Reed, P., Zakhari, S., Read, J., Watson, B. and Guyeret, M. 2009. The NIH human microbiome project. Genome. Res. 19, 2317–2323. Puente, E., Alonso, L., Laganà, A.S., Ghezzi, F., Casarin, J. and Carugno, J. 2020. Chronic endometritis: old problem, novel insights and future challenges. Int. J. Fertil. Steril. 13, 250–256. Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., Peplies, J., Ludwig, W. and Glöckner, F.O. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic. Acids. Res. 41, 590–596. Rodionova, I.A., Scott, D.A., Grishin, N.V., Osterman, A.L. and Rodionov, D.A. 2012. Tagaturonate–fructuronate epimerase UxaE, a novel enzyme in the hexuronate catabolic network in Thermotoga marítima. Environ. Microbio. 14, 2920–2934. Santibáñez, R., Lara, F., Barros, T., Mardones, E., Cuadra, F. and Thomson, P. 2022. Ocular microbiome in a group of clinically healthy horses. Animals 712, 943. Sathe, S., Leiken, A. and Plummer, P. 2017. Metagenomic sequencing of the uterine microbial environment during estrus and early pregnancy in mares. Clin. Theriogenol. 9, 453. Schnobrich, M., Atwood, K., Barr, B., Bradecamp, E. and Scoggin, C. 2017. Sequencing, culture and cytology results in 10 clinically normal mares. Clin. Theriogenol. 9, 443. Shanahan, F., Ghosh, T.S. and O’Toole, P.W. 2021. The healthy microbiome—what is the definition of a healthy gut microbiome?. Gastroenterology 160, 483–494. Tessier, H. 1900. Recherches sur la flore intestinale des nourrissons (état normal et pathologique). Thesis of doctorade, Paris, pp: 1–253. Torraca, V., Holt, K. and Mostowy, S. 2020. Shigella sonnei. Trends. Microbiol. 28, 696–697. Troedsson, M.H.T. and Woodward, E.M. 2016. Our current understanding of the pathophysiology of equine endometritis with an emphasis on breeding-induced endometritis. Reprod. Biol. 16, 8–12. Vaneechoutte, M., Devriese, L.A., Dijkshoorn, L., Lamote, B., Deprez, P., Verschraegen, G. and Haesebrouck, F. 2000. Acinetobacter baumannii-infected vascular catheters collected from horses in an equine clinic. J. Clin. Microbiol. 38, 4280–4281. Wang, Q., Garrity, G.M., Tiedje, J.M. and Cole, J.R. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. Yang, X., Cheng, G., Li, C., Yang, J., Li, J., Chen, D., Zou, W., Jin, S.Y., Zhang, H., Li, D., He, Y., Wang, C., Wang, M. and Wang, H. 2017. The normal vaginal and uterine bacterial microbiome in giant pandas (Ailuropoda melanoleuca). Microbiol. Res. 199, 1–9. Yilmaz, P., Parfrey, L.W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., Schweer, T., Peplies, J., Ludwig, W. and Glöckner, F.O. 2014. The SILVA and “all-species living tree project (LTP)” taxonomic frameworks. Nucleic. Acids. Res. 42, 643–648. | ||
| How to Cite this Article |
| Pubmed Style Thomson P, Pareja J, AN, Santibáñez R, Castro R. Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares. Open Vet J. 2022; 12(6): 797-805. doi:10.5455/OVJ.2022.v12.i6.3 Web Style Thomson P, Pareja J, AN, Santibáñez R, Castro R. Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares. https://www.openveterinaryjournal.com/?mno=26153 [Access: July 27, 2024]. doi:10.5455/OVJ.2022.v12.i6.3 AMA (American Medical Association) Style Thomson P, Pareja J, AN, Santibáñez R, Castro R. Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares. Open Vet J. 2022; 12(6): 797-805. doi:10.5455/OVJ.2022.v12.i6.3 Vancouver/ICMJE Style Thomson P, Pareja J, AN, Santibáñez R, Castro R. Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares. Open Vet J. (2022), [cited July 27, 2024]; 12(6): 797-805. doi:10.5455/OVJ.2022.v12.i6.3 Harvard Style Thomson, P., Pareja, . J., , . A. N., Santibáñez, . R. & Castro, . R. (2022) Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares. Open Vet J, 12 (6), 797-805. doi:10.5455/OVJ.2022.v12.i6.3 Turabian Style Thomson, Pamela, Josefina Pareja, Andrea Núñez, Rodrigo Santibáñez, and Rodrigo Castro. 2022. Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares. Open Veterinary Journal, 12 (6), 797-805. doi:10.5455/OVJ.2022.v12.i6.3 Chicago Style Thomson, Pamela, Josefina Pareja, Andrea Núñez, Rodrigo Santibáñez, and Rodrigo Castro. "Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares." Open Veterinary Journal 12 (2022), 797-805. doi:10.5455/OVJ.2022.v12.i6.3 MLA (The Modern Language Association) Style Thomson, Pamela, Josefina Pareja, Andrea Núñez, Rodrigo Santibáñez, and Rodrigo Castro. "Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares." Open Veterinary Journal 12.6 (2022), 797-805. Print. doi:10.5455/OVJ.2022.v12.i6.3 APA (American Psychological Association) Style Thomson, P., Pareja, . J., , . A. N., Santibáñez, . R. & Castro, . R. (2022) Characterization of microbial communities and predicted metabolic pathways in the uterus of healthy mares. Open Veterinary Journal, 12 (6), 797-805. doi:10.5455/OVJ.2022.v12.i6.3 |

