




| Original Article | ||
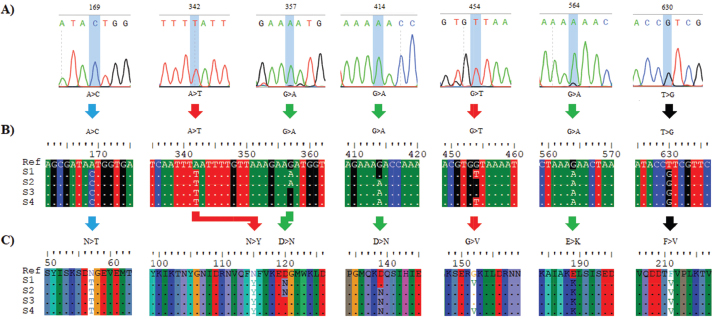
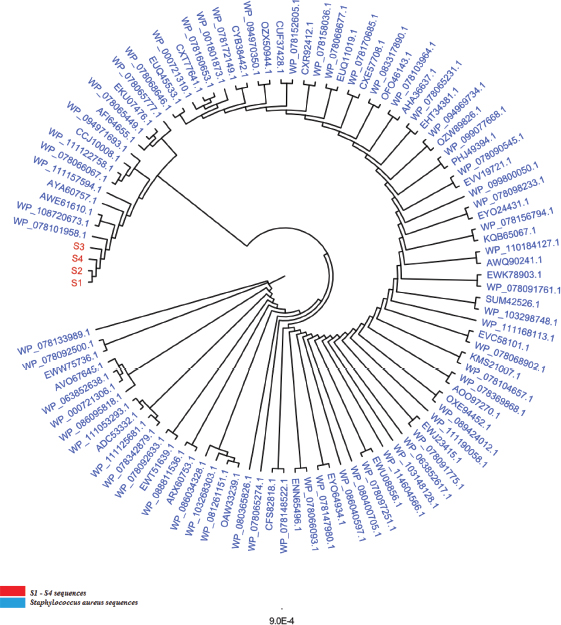
Open Vet J. 2019; 9(4): 339-348 doi: 10.4314/ovj.v9i4.12 Open Veterinary Journal, (2019), Vol. 9(4): 339–348 Original Research DOI: http://dx.doi.org/10.4314/ovj.v9i4.12 The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolatesSarhan R. Sarhan1, Hayder O. Hashim2 and Mohammed Baqur S. Al-Shuhaib3*1Department of Pharmacology and Physiology, College of Veterinary Medicine, Wasit University, Wasit, Iraq 2Department of Clinical Laboratory Sciences, College of Pharmacy, University of Babylon, Iraq 3Department of Animal Production, College of Agriculture, Al-Qasim Green University, Al-Qasim, Babil 51001, Iraq *Corresponding Author: Mohammed Baqur S. Al-Shuhaib. Department of Animal Production, College of Agriculture, Al-Qasim Green University, Al-Qasim, Babil 51001, Iraq. Email: mohammed79 [at] agre.uoqasim.edu.iq Submitted: 22/05/2019 Accepted: 21/11/2019 Published: 22/12/2019 © 2019 Open Veterinary Journal AbstractBackground: The mecA gene is a key factor that allows bacterial cells to resist several antibiotics. Aim: This study was conducted to detect the mecA gene polymorphism in ovine wounds and its possible association with the structure and function of penicillin binding protein A2 (PBP2A). Methods: One genetic locus of 1,967 bp that covered the majority of the coding regions of the mecA gene within methicillin-resistant Staphylococcus aureus (MRSA) DNA sequences was designed. Results: In addition to standard microbiological tests, PCR-sequencing reactions and phylogenetic analyses confirmed the identity of the targeted MRSA bacteria. Seven novel missense SNPs, including N57T, N115Y, D120N, D139N, G152V, E189K, and F211V, were observed in the mecA amplicons. Multiple state-of-the-art in silico tools were utilized to assess the consequences of each observed SNP in terms of its effect on the corresponding PBP2A protein structure and function. It was shown that some MRSA isolates exhibited a highly PBP2A-damaging SNP, G152V, which showed an entirely deleterious effect on the PBP2A. Furthermore, G152V induced an alteration in the PBP2A interaction with its receptor, which presumably reduced its affinity to bind with the beta-lactams. Conclusion: The present report indicated a possible role for the observed deleterious G152V SNP in the reduction of PBP2A binding with beta-lactams, which has led to a remarkable increase in MRSA’s resistance to antibiotics. Keywords: Infection, In silico, MecA, Missense, Variations. IntroductionMethicillin-resistant Staphylococcus aureus (MRSA) is a serious cause of health-care associated infections. MRSA develops resistance to a large group of antibiotics called beta-lactams, such as penicillin and cephalosporins, tetracyclines, and daptomycin, which are usually prescribed for many Gram-positive, Gram-negative, and anaerobic organisms (Choo and Chambers, 2016). Although S. aureus is a commensal to its animal host (Jenkins et al., 2015), it may become serious if developed antimicrobial resistance (Monecke et al., 2011). There has been a steady increase in the prevalence of MRSA all over the world (Kumar et al., 2015). MRSA strains have gradually emerged as major pathogens because of their potential to acquire resistance toward many classes of antimicrobial chemotherapeutic agents (Kaur and Chate, 2015). In recent years, MRSA has been identified as an important pathogen in livestock and several domesticated species (Dahms et al., 2014; Smith, 2015). Similarly, MRSA strains have equally adopted as major pathogens of public health concern because of their capacity to adapt to different environmental conditions (Boswihi and Udo, 2018). Although the majority of studies have shown that the numbers of MRSA strains have increased remarkably due to the quick loss of beta-lactams therapeutic ability to confront these pathogenic bacteria (Virdis et al., 2010), beta-lactam antibiotics still the treatment of choice for staphylococcal infections and the administration of methicillin represents one of the therapeutic means for treatment of many cases of S. aureus infection (Hryniewicz and Garbacz, 2017). However, such resistance to beta-lactams is mediated through an altered protein called penicillin binding protein (PBP2A), with reduced affinity for beta-lactams antibiotics, and, therefore, not inhibited by antibiotics treatment (Makgotlho et al., 2009). The staphylococcal PBPs are transpeptidases that are involved in the transport of peptide precursors from the cytoplasm to the cell membrane. Modified PBP2A has a low affinity for beta-lactams. Therefore, due to the presence of the new altered protein, methicillin-resistant strains are no longer susceptible to beta-lactams, and they exhibit resistance to antibiotics from many other groups (Appelbaum, 2007). The PBP2A is encoded by the mecA gene, which is carried on a 21- to 67-kb element in a chromosomal mobile genetic element known as Staphylococcal cassette chromosome mec (SCCmec) (Grundmann et al., 2006). Due to a possible association of MRSA with multiple antibiotic resistance, the accurate and rapid identification of MRSA is crucial in the clinical world for the timely management of bacterial infections (Johnson, 2011). Accordingly, MRSA has become one of the most commonly diagnosed causes of skin and soft-tissue infections, including wounds, burns, and abscesses (Yahaghi et al., 2014). Therefore, rapid recognition and detection of the MRSA strains are essential for promoting effective therapy, preventing the distribution of infection, and reducing the risk of patient mortality (Torimiro and Torimiro, 2012). Although the absolute detection of the mecA gene in all MRSA isolates is being questioned (Elhassan et al., 2015), the identification of S. aureus through PCR amplification of the mecA gene is considered as a standard gold method for rapid detection of the majority of MRSA infections (Anand et al., 2009). However, few studies had highlighted the occurrence of MRSA in sheep (Petinaki and Spiliopoulou, 2012). Similarly, no sufficient light has been focused on the mecA genetic polymorphisms’ effects in the final manifestation of PBP2A. In this regard, we conduct the present investigation to assess the occurrence of MRSA isolated from wound samples collected from Awassi sheep, to provide in vitro molecular characterization of MRSA isolates as well as to assess its in silico consequences on the structure and function of PBP2A, with a special emphasis on its binding with the beta-lactams. Materials and MethodsSamples collectionThe research was conducted by the main guidelines mentioned in the international recommendations for the care and use of animals, and all the samples were collected from sheep by proper animal welfare guidelines for the care and use of animals (Federation of Animal Science Societies, 2010). A total of 60 non-repeated pus/wound swap samples from anatomically-identified wounds in the Iraqi Awassi sheep breed aged 3–5 years in one grazing region within Wasit governorate was investigated for the presence of S. aureus. The included sheep population had been intermittently treated by several beta-lactams for extended periods until their breeders noticed a reduced recovery in this breed due to bacterial infection and, therefore, asked for the intervention of veterinarians to solve this issue. Superficial samples of wound swabs were collected from the infected tissues of each animal and were inoculated on mannitol salt agar and blood agar (Oxoid, UK) and incubated at 37°C for 48 hours. Isolation and identification of S. aureusThe culture characteristics, based on colonial growth morphology, Gram stain, and several biochemical tests, were performed for the isolation and identification of S. aureus, including β-hemolysis on blood agar, catalase, urease, oxidase, and DNase (Forbes et al., 2007). The isolates of S. aureus were subjected to several beta-lactam antibiotics, including cefoxitin, gentamycin, oxacillin, and penicillin G Discs (Bioanalyse, Turkey), using a 30, 10 μg, 5, and 10 μg disc, respectively, to test the presence of MRSA. A 0.5 McFarland standard suspension of the isolate was made, and lawn culture was done on a Muller-Hinton agar plate (Oxoid, UK). Plates were incubated at 37°C for 18 hours, and zone diameters were measured according to the Clinical and Laboratory Standard Institute (CLSI, 2011). DNA extractionDNA samples were extracted from S. aureus cultures using a bacterial genomic DNA extraction kit, according to the manufacturer’s instructions (Cat #GBB100/101, Geneaid Biotech, Taiwan). The concentration and purity of DNA were measured by a nanodrop (BioDrop μLITE, BioDrop, UK), while the DNA integrity was checked by a standard 0.8% (w/v) agarose gel electrophoresis that was pre-stained with a higher concentration of ethidium bromide (0.7 μg/ml) in TAE (40 mM Tris-acetate; 2 mM EDTA, pH 8.3) buffer. The isolated DNA was used as a template for the mecA gene to confirm the identification of S. aureus. PCRA PCR fragment covered 1,967 bp within the coding portion of the mecA gene within the S. aureus genome was designed using the NCBI-primer blast tool to accommodate the largest possible amplicons (Ye et al., 2012). The lyophilized primers (mecA-F; 5′- TGAAAAAGATAAAAATTGTTCCACT-3′ and mecA-R; 5′- TTTACCTGAGATTTTGGCATTG -3′) were purchased from Bioneer (Bioneer, Daejeon, South Korea). The PCR reaction was performed with a high fidelity AccuPower pfu PCR premix (Cat # K-2022, Bioneer, Daejeon, South Korea). Each PCR premix contained 2.5 U of pfu DNA polymerase, 250 μM of dNTPs, 10 mM of Tris-HCl (pH 9.0), 30 mM of KCl, 1.5 mM of MgCl2. The reaction mixture was completed with 1 μl of 10 μM of forward and reverse primers and 50 ng of genomic DNA. The following program was applied in a PCR thermocycler (Bioneer, Daejeon, South Korea). The amplification began with the initial denaturation at 94°C for 5 minutes, followed by 30 cycles of denaturation at 94°C, annealing at 58°C, and elongation at 72°C for 1 minute each, and was finalized with a final extension at 72°C for 10 minutes. Amplification was verified by electrophoresis on an ethidium bromide (0.5 μg/ml) pre-stained 1.5% (w/v) agarose gel in 1× TBE buffer (2 mM of EDTA, 90 mM of Tris-Borate, pH 8.3). DNA sequencingThe resolved PCR amplicons were commercially sequenced from both forward and reverse termini, according to the instruction manual of the sequencing company (Macrogen Inc. Geumchen, Seoul, South Korea). Only clear chromatographs obtained from ABI sequence files were further analyzed, ensuring that the annotation and variations are not because of PCR or sequencing artifacts. By comparing the observed DNA sequences of the isolated bacterial samples with the retrieved DNA sequences of S. aureus strain MRSA, the virtual positions and other details of the retrieved PCR fragments were identified. The sequencing results of the PCR products of different samples were edited, aligned, and analyzed along with the respective sequences in the reference database using BioEdit Sequence Alignment Editor Software Version 7.1 (DNASTAR, Madison, WI). The observed variations in each sequenced sample were numbered in PCR amplicons as well as in its corresponding position within the reference genome. Construction of phylogenetic treeA specific comprehensive bacterial tree was constructed in which the observed bacterial variants were compared with their neighbor homologous bacterial sequences, using a neighbor joining method described by Al-Shuhaib et al. (2018a). Next, the blast results of the observed variants were combined and aligned together using Clustal Omega-based tools. A full inclusive tree, including the observed variant, was visualized as a polar cladogram, using the Figtree tool (http://tree.bio.ed.ac.uk/software/figtree/). The bacterial sequences of each classified phylogenetic species-group were colored appropriately according to the identity of the included organism within the comprehensive tree (Al-Dabbagh et al., 2019). In silico computationAll observed missense SNPs were prioritized, using several computational tools to assess their effect on PBP2A. The SIFT program was utilized to assess the structural effect of SNP on the PBP2A (Pauline and Steven, 2003). The functionality of each SNP was annotated using the PolyPhen-2 tool (Adzhubei et al., 2010). Similarly, the biological impact of the observed missense SNP on the resulting protein was predicted by PROVEAN software (Choi et al., 2012), while the severity of nsSNPs was assessed using SNAP2 tool (Smigielski et al., 2000). The role of each amino acid substitution in the post-translational modifications, including glycation, phosphorylation, ubiquitylation, and sumoylation of PBP2A was predicted through NetGlycate1.0 (Johansen et al., 2006), NetPhos3.0 (Blom et al., 2004), UbPerd (Radivojac et al., 2010), and SUMOplot (http://www.abgent.com/sumoplot), respectively. The 3-D structure of the PBP2A was constructed from the RaptorX software (Källberg et al., 2012). The virtual proposed changes within its corresponding mutants were performed with PyMOL-v1, 7.0.1 software (www.shrodinger.com). Subsequently, the conservation status of the observed SNPs was assessed by a ConSurf machine (Ashkenazy et al., 2010). The potential role of missense SNPs in the binding with the receptor was predicted by FTsite server (Kozakov et al., 2015). Ethical approvalAll animal handling procedures were in accordance with the Regulations of the standard animal’s regulation care. The experimental protocol was approved by The Scientific Ethics Committee of the University of Babylon. ResultsStandardized laboratory diagnostic tests showed that S. aureus isolates were found in 40% of the total bacterial infections in wound samples (24 S. aureus /60 wound samples). Our finding revealed that about 41.6% (10/24 S. aureus samples) of identified S. aureus isolates were MRSA since they had shown moderate to high resistance to all antibiotics used, including penicillin, cefoxitin, and oxacillin, while the residual 14 isolates did not show any detectable resistance. However, the subsequent downstream PCR experiments of the mecA amplicons were performed for all 24 S. aureus isolates. PCR results indicated the absence of the mecA gene in all antimicrobial-sensitive 14 isolates, while the presence of the mecA amplicons was confirmed for all 10 MRSA isolates. The alignment results of the specific 1967 bp amplicons revealed the detection of seven mutations in the analyzed samples compared to the deposited mecA GenBank sequences. The sequencing chromatogram of each observed substitution mutation, as well as its detailed annotations, was documented, as shown in Figure 1A. Sequencing results of the specific 1,967 bp amplicons revealed the detection of seven mutations in the analyzed samples compared to the deposited mecA GenBank sequences. The sequencing chromatogram of each observed substitution mutation, as well as its detailed annotations, was documented as shown in Figure 1A. The DNA sequence alignment of the observed variations detected by a direct PCR-sequencing method was shown in Figure 1B. The alignment of the translated sequences indicated the observation of seven missense SNPs. All of these nsSNPs were variably distributed in the coding region of the mecA gene (Fig. 1C). All detected mecA gene variations were deposited in the NCBI reference sequences (MK034953, MK034954, MK034955, and MK034956 for the observed S1–S4 variants, respectively). A downstream phylogenetic analysis was performed to identify the precise position of the studied isolated in terms of the phylogenetic positioning within related bacterial species to add another layer of confirmation about the assured identity of S. aureus isolates. The total number of the aligned nucleic acid sequences, irrespective of the bacterial variants, in this comprehensive tree was 100 sequences. The constructed comprehensive tree indicated the presence of only one species throughout the 100 scanned mecA variant sequence—related species, namely, S. aureus (Fig. 2). The phylogenetic analysis had exhibited an extremely high S. aureus detection specificity concerning mecA-based phylogenetic protocols, which provide an inclusive tool regarding the high ability of such designed mecA-based genetic fragment to identify S. aureus isolates efficiently.
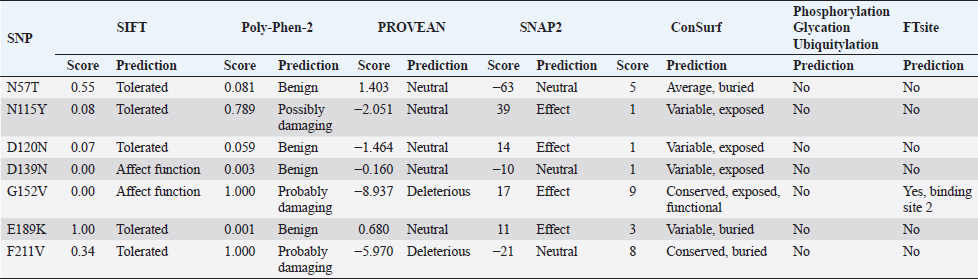
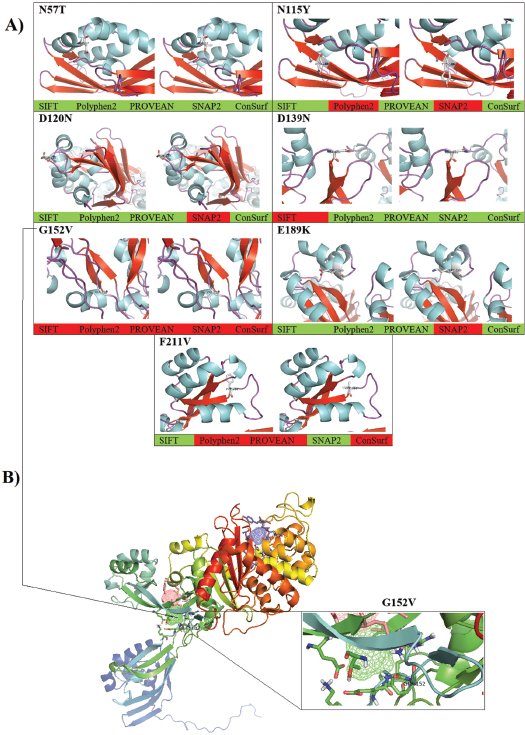
Fig. 1. The detected variations in the mecA gene within the methicillin-resistance Staphylococcus aureus isolates. (A) Chromatograms of the observed nucleic acid substitutions in the 1,967 bp amplicons. (B) Multiple DNA sequences alignment. (C) Multiple protein sequences alignment. The symbol “>” refers to a substitution mutation, “ref” refers to “reference sequences”, S1 refers to isolates 1, and 2, S2 refers to isolates 3, 4, and 5, S3 refers to isolates 6, and 8, S4 refers to samples 7, 9, and 10. Picture of branch A was generated by Snap gene viewer, while pictures of branches B and C were generated by Edit Seq Lasergene software. As long as all the observed seven SNPs had exhibited a missense effect, such dramatic changes in amino acid residues may have several consequences on protein structure, function, or both. Therefore, several in silico tools were utilized in this contest to explore the role of these mutations in the subsequent potential alteration in the resulting protein. The observed missense SNPs were further analyzed to identify whether they substitute the corresponding position in the mecA gene-encoded PBP2A. The consequences of these nsSNPs were evaluated using different, publicly available, computational algorithms. By comparing the prediction of these methods, the non-deleterious to deleterious consequences of these nsSNPs were confirmed in the utilized tools collectively. Thus, the validation of such effects was provided through several other tools, such as SIFT, PolyPhen-2, PROVEAN, SNAP2, and ConSurf tools, respectively (Table 1). These computational programs have shown some discrepancies in the results, as revealed in N115Y, D120N, D139N, E189K, and F211V. More in-depth computational data was provided through constructing a virtual 3-D structure of the PBP2A. The graphical 3-D representation of the PBP2A had shown virtual status for each observed variant. However, G152V was the most deleterious one, as all cumulative in silico tools had indicated the entire deleterious consequences for this SNP. The cumulative in silico computation of the deleterious effect of each SNP had predicted several forms of damaging effects that started from G152V as being the most deleterious SNP, which followed by F211V by taking a less damaging effect. Subsequently, other SNPs had less deleterious consequences, including N115Y, E189K, and D120N. Finally, an entirely non-deleterious SNP was shown cumulatively, which was N57T (Fig. 3A). Moreover, several in silico post-translational modification prediction tools, such as NetGlycate1.0, NetPhos3.0, UbPerd, and SUMOplot were collectively employed to identify the potential participation of the observed missense SNPs in the post-translational modification of PBP2A. No involvement was observed for the observed nsSNP in all studied post-translational glycation, phosphorylation, ubiquitylation, and sumoylation modification sites. The FTsite program was usually implemented to predict whether each amino acid being mutated is positioned in a ligand binding site in the protein of interest. The FTsite had indicated that there was no potential ligand binding site for the observed nsSNP, including N57T, N115Y, D120N, D139N, E189K, and F211V, while the most deleterious G152V had exerted another dramatically modifying effect, in terms of binding with the receptors (Fig. 3B). The G152V-induced PBP2A alteration was the only receptor modulation effect that exhibited from the observed SNPs. DiscussionBefore exploring the genetic polymorphism of MRSA strains, it is necessary to confirm the phylogenetic identity of the isolated S. aureus by comparing the observed PBP2A variants with its neighbor sequences. However, the obvious identity of the currently analyzed MRSA strains varieties is determined from constructing an inclusive phylogenetic tree for all four PBP2A variations. Despite this unique characterization of the studied PBP2A variants, no deviation from S. aureus species was noticed concerning all phylogenetically analyzed variants. Although these isolates exhibited a particular position away from other isolates, no distinct deviation was from each other as all observed PBP2A S1–S4 groups were localized in only one particular position within this comprehensive tree. As long as PCR-sequencing, this phylogenetic notion provided a further indication of the identity of these wound-infecting isolates. However, this pattern of phylogenetic distribution may refer to a certain evolutionary role that may be conserved in these isolates to represent a particular mode of sequence variations among them. The genetic biodiversity of MRSA could not be eliminated from this aspect. Furthermore, specific prioritizing roles of in silico tools have been shown through analyzing the range of the damaging effect of each SNP. However, several differences in the prediction results were detected. This observation is attributed to the difference in the algorithm that each software was relied on in the prediction. Therefore, several differences in the outcomes were expected to occur at some points (Al-Shuhaib et al., 2018b). Therefore, the cumulative prediction has provided a clear prioritizing tool for the observed SNPs, by showing a series of deleterious effects of the observed SNPs to assess their grade of the effect of the altered proteins. This action would enhance the way of our understanding of the nature of polymorphism in this pathogenic bacterial isolates. The presence of the highly deleterious G152V only in S1 and S4 bacterial groups and the non-deleterious N57T in all examined samples has indicated the limited deleterious effect on the examined bacterial isolates. One of the well-known mechanisms for methicillin resistance in MRSA isolates has been reported to arise from the expression of altered forms of PBP2A that has a lower penicillin binding affinity with the drug compared to the normal PBP2A (Stapleton and Tylor, 2002). However, it could be rational to state that both S1 and S4 pathogenic bacterial species have a remarkable activity in the wound region and not significantly affected by any medical treatments. This observation entails a steady development of methicillin resistance strategy that arises from continuous treatment with beta-lactams (Shaikh et al., 2015). Meanwhile, the other observed nsSNPs have not revealed such a deleterious manner, as it was predicted cumulatively. However, the currently utilized prediction tools have prioritized the observed SNPs into four main categories the highly deleterious class, as in G152V; the moderately deleterious nsSNP, as in F211V and N115Y; the low deleterious class, as in D120N, D139N, and E19K; and the non-deleterious class, as in N57T. Although the commonly used in silico tools have helped us determine the clear damaging effect of G152V on the PBP2A, they do not give us a clear indication about the role of these SNPs in the resistance of MRSA for beta-lactam drugs. Therefore, a further explanation for this damaging role has been suggested by the FTsite tool (Kozakov et al., 2015). The latter tool has clearly indicated a dramatic modulation of PBP2A binding with the receptor in the G-152 residue position. This modulation has suggested an alteration in the binding with the beta-lactam antibiotics molecules upon mutation with this SNP. Therefore, the currently identified G152V might be associated with the possible resistance of MRSA strains to the commonly used beta-lactams. The FTsite observation has confirmed this postulated strategy by indicating a possible alteration of G152V-mutant PBP2A for the binding with receptors, which negatively affect the ability of this protein to adhere to beta-lactams. Therefore, in addition to the well accepted view regarding MRSA antibiotics resistance (Aedo and Tomasz, 2016), the currently used in silico computations have provided a conceivable insight into the role of the genetic polymorphism strategy of MRSA to protect itself from the beta-lactam attack. The presence of the interesting G152V role in this strategy has provided an easy prediction for the direct involvement of this deleterious SNP in preventing the binding of the mutant MRSA bacterial strains with administrated antibiotics on their cell wall. Furthermore, the G152V role in the PBP2A receptor binding alteration may suggest that S1 and S4 MRSA strains have developed a particular genetic mechanism to overcome the binding with the therapeutic beta-lactam agents. Although an accumulated number of reports have indicated the ability of MRSA to resist antibiotics through altering PBP2A (Foster, 2017), only one study referred to the presence of two amino acid substitutions, Y446N and E447K, in PBP2A that are responsible for such S. aureus resistance in MRSA strains isolated from patients with cystic fibrosis (Long et al., 2014). Meanwhile, the present study has suggested a putative mechanism mediated by G152V to explain this resistance in MRSA strains isolated from infected sheep. Although all the sequenced MRSA strains were found to exert a full beta-lactam resistance, G152V was not found in all sequenced MRSA strains. This notion may be associated with the ability of some MRSA strains to counterfeit the action of antibiotics by this strategy, while other MRSA strains, such as S2 and S3, have rather developed another resistance strategy by relying upon unknown mechanisms.
Fig. 2. The comprehensive phylogenetic tree of genetic variants of the mecA genetic fragments for the wound-infecting Staphylococcus aureus isolates. The red color refers to the sequenced S1–S4 variants, while the cyan color refers to the related reference NCBI Staphylococcus aureus deposited species. All the mentioned numbers referred to Genbank acc. no. of each referring species. The number “9.0” at the bottom of the tree refers to the degree of scale range among the comprehensive tree categorized organisms. S1 refers to isolates 1, and 2, S2 refers to isolates 3, 4, and 5, S3 refers to isolates 6, and 8, S4 refers to samples 7, 9, and 10. Table 1. The in silico analysis of the observed nonsynonymous SNPs in PBP2A using the commonly available bioinformatics tools to shows their potential effects and their potential post-translation interactions.
Fig. 3. The computational prediction of the observed variations in the PBP2A within Staphylococcus aureus strain MRSA. (A) The cumulative in silico prediction of N57T, N115Y, D120N, D139N, G152V, E189K, and F211V. The grade of the deleterious effect was shown, in which the red color refers to the deleterious effect of the SNP, while the green color refers to the neutral effect of the same SNP. (B) The predictive binding sites of PBP2A using the FTsite program. The highlighted position is the Gly-152, in which a remarkable change in the binding with the receptor is taken place. In conclusion, the present study describes for the first time that MRSA strains developed resistance to beta-lactams through modulating the receptor-binding site of their PBP2A proteins to prevent any further interaction between this protein and the administrated antibiotics on the cell wall MRSA of the treated animals. The alteration in the binding with the PBP2A receptor was not the only predicted mechanism by the utilized prediction tools; instead, the highly deleterious role of G152V effects was also predicted. As currently determined cumulatively, this extremely deleterious consequence may be involved in reducing the activity of PBP2A itself without being focused on the direct binding with the beta-lactam antibiotics. These tools may exhibit an additional reducing effect on the resulting PBP2A, which may represent a second putative strategy through which the manifested protein is negatively affected by missense SNP. Eventually, whatever the mechanism through which G152V affects the altered protein, the mutant protein is predictively unable to bind with the beta-lactams, which is the real point behind this computational prediction. The resulting low-binding affinity between PBP2A and beta-lactams on the mutant MRSA isolates may prevent PBP2A-mediated cell wall disruption, which leads to more resistance of the mutant MRSA isolates to beta-lactam drugs. AcknowledgmentsThis study was supported by Alfadhil Scientific training company, Babil, Iraq (018-2). Conflict of interestThe authors declare that there is no conflict of interest. Authors’ contributionsS.H.S. involved in all sampling and experimental procedures of the manuscripts; H.O.H. conceived the main concept of the study and involved in some experimental procedures. M.B.S.A. analyzed data and wrote the manuscript. ReferencesAdzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S.R. 2010. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. Aedo, S. and Tomasz, A. 2016. Role of the stringent stress response in the antibiotic resistance phenotype of methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 25(60), 2311–2317. Al-Dabbagh, N.N., Hashim, H.O. and Al-Shuhaib, M.B.S. 2019. A highly efficient computational discrimination among Streptococcal species of periodontitis patients using 16S rRNA amplicons. Korean J. Microbiol. 55(1), 1–8. Al-Shuhaib, M.B.S., Al-Kaaby, H.N. and Alwan, S.L. 2018a. A highly efficient electrophoretic method for discrimination between two Neoscytalidium species using a specific fungal internal transcribed spacer (ITS) fragment. Folia Microbiol. 64(2), 161–170. Al-Shuhaib, M.B.S., Al-Kafajy, F.R., Badi, M.A., AbdulAzeez, S., Marimuthu, K., Al-Juhaishi, H.A.I. and Borgio, J.F. 2018b. Highly deleterious variations in COX1, CYTB, SCG5, FK2, PRL, and PGF genes are the potential adaptation of the immigrated African ostrich population. Comput. Biol. Med. 100, 17–26. Anand, K.B., Agrawal, P., Kumar, S. and Kaplia, K. 2009. Comparison of cefoxitin disc diffusion test, oxacillin screen agar, and PCR for mecA gene for detection of MRSA. Indian J. Med. Microbiol. 27, 27–29. Appelbaum, P.C. 2007. Microbiology of antibiotic resistance in Staphylococcus aureus. Clin. Infect. Dis. 45, S165–S170. Ashkenazy, H., Erez, E., Martz, E., Pupko, T. and Ben-Tal, N. 2010. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533. Blom, N., Sicheritz-Ponten, T., Gupta, R., Gammeltoft, S. and Brunak, S. 2004. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649. Boswihi, S.S. and Udo, E.E. 2018. Methicillin-resistant Staphylococcus aureus: An update on the epidemiology, treatment options and infection control. Curr. Med. Res. Pract. 8, 18–24. Choi, Y., Sims, G.E., Murphy, S., Miller, J.R. and Chan, A.P. 2012. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7, e46688. Choo, E.J. and Chambers, H.F. 2016. Treatment of Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect. Chemother. 48, 267–273. CLSI, 2011. Performance standards for antimicrobial disk and dilution susceptibility testing procedures. Document M 100-S21 ed. Wayne, PA. Dahms, C., Hübner, N.O., Cuny, C. and Kramer, A. 2014. Occurrence of methicillin-resistant Staphylococcus aureus in farm workers and the livestock environment in Mecklenburg-Western Pomerania, Germany. Acta Vet. Scand. 56, 53. doi:10.1186/s13028-014-0053-3 Elhassan, M.M., Ozbak, H.A., Hemeg, H.A., Elmekki, M.A. and Ahmed, L.M. 2015. Absence of the mecA Gene in Methicillin Resistant Staphylococcus aureus Isolated from Different Clinical Specimens in Shendi City, Sudan. Biomed. Res. Int. 2015, 895860. doi:10.1155/2015/895860 Federation of Animal Science Societies. 2010. Guide for the care and use of agricultural animals in research and teaching, 3rd ed. Champaign, IL: 61822. Forbes, B., Sahm, D.F. and Weissfeld, A.S. 2007. Bailey and Scott’s diagnostic microbiology, 12th ed. MO: Mosby Inc, Maryland Heights. Foster, T.G. 2017. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 41, 430–449. Grundmann, H., Aires-de-Sousa, M., Boyce, J. and Tiemersma, E. 2006. Emergence and resurgence of methicillin-resistant Staphylococcus aureus as a public threat. The Lancet 368, 874–885. Hryniewicz, M.M. and Garbacz, K. 2017. Borderline oxacillin-resistant Staphylococcus aureus (BORSA) – a more common problem than expected? J. Med. Microbiol. 66, 1367–1373. Jenkins, A., Diep, B.A., Mai, T.T., Vo, N.H., Warrener, P., Suzich, J., Stover, C.K. and Sellman, B.R. 2015. Differential expression and roles of Staphylococcus aureus virulence determinants during colonization and disease. MBio. 6, e02272–14. doi:10.1128/mBio.02272-14 Johansen, M.B., Kiemer, L. and Brunak, S. 2006. Analysis and prediction of mammalian protein glycation. Glycobiology 16, 844–853. Johnson, A.P. 2011. Methicillin-resistant Staphylococcus aureus: the European landscape. J. Antimicrob. Chemother. 66(S4), iv43–iv48. Källberg, M., Wang, H., Wang, S., Peng, J., Wang, Z., Lu, H. and Xu, J. 2012. Template-based protein structure modeling using the RaptorX web server. Nat. Protoc. 7, 1511–1522. Kaur, D.C. and Chate, S.S. 2015. Study of antibiotic resistance pattern in methicillin resistant Staphylococcus aureus with special reference to newer antibiotic. J. Glob. Infect. Dis. 7, 78–84. Kozakov, D., Grove, L.E., Hall, D.R., Bohnuud, T., Mottarella, S.E., Luo, L., Xia, B., Beglov, D. and Vajda, S. 2015. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 10, 733–755. Kumar, N., David, M.Z., Boyle-Vavra, S., Sieth, J. and Daum, R.S. 2015. High Staphylococcus aureus colonization prevalence among patients with skin and soft tissue infections and controls in an urban emergency department. J. Clin. Microbiol. 53, 810–815. Long, S.W., Olsen, R.J., Mehta, S.C., Palzkill, T., Cernoch, P.L., Perez, K.K., Musick, W.L., Rosato, A.E. and Musser, J.M. 2014. PBP2a mutations causing high-level Ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 58, 6668–6674. Makgotlho, P.E., Kock, M.M., Hoosen, A., Lekalakala, R., Omar, S., Dove, M. and Ehlers, M.M. 2009. Molecular identification and genotyping of MRSA isolates. FEMS Immunol. Med. Microbiol. 57, 104–115. Monecke, S., Coombs, G., Shore, A.C., Coleman, D.C., Akpaka, P., Borg, M., Chow, H., Ip, M., Jatzwauk, L., Jonas, D., Kadlec, K., Kearns, A., Laurent, F., O'Brien, F.G., Pearson, J., Ruppelt, A., Schwarz, S., Scicluna, E., Slickers, P., Tan, H.L., Weber, S. and Ehricht, R. 2011. A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS One 6, e17936. doi:10.1371/journal.pone.0017936 Pauline, C.N. and Steven, H. 2003. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814. Petinaki, E. and Spiliopoulou, I. 2012. Methicillin-resistant Staphylococcus aureus among companion and food-chain animals: impact of human contacts. Clin. Microbiol. Infec. 18, 626–634. Radivojac, P., Vacic, V., Haynes, C., Cocklin, R.R., Mohan, A., Heyen, J.W., Goebl, M.G. and Iakoucheva, L.M. 2010. Identification, analysis, and prediction of protein ubiquitination sites. Proteins 78(2), 365–380. Shaikh, S., Fatima, J., Shakil, S., Mohd, S., Rizvi, D. and Kamal, M.A. 2015. Antibiotic resistance and extended spectrum beta-lactamases: Types, epidemiology and treatment. Saudi J. Biol. Sci. 22, 90–101. Smigielski, E.M., Sirotkin, K., Ward, M. and Sherry, S.T. 2000. dbSNP: a database of single nucleotide polymorphisms. Nucleic Acids Res. 28, 352–355. Smith, T.C. 2015. Livestock-Associated Staphylococcus aureus: The United States Experience. PLoS Pathog. 11, e1004564. doi:10.1371/journal.ppat.1004564. Stapleton, P.D. and Taylor, P.W. 2002. Methicillin resistance in Staphylococcus aureus: mechanisms and modulation. Sci. Prog. 85, 57–72. Torimiro, N. and Torimiro, S.E.A. 2012. Antibiotic resistance profile in community-associated Staphylococcus aureus strains isolated from a Nigerian peri-urban community. African J. Biotechnol. 11, 16071–16076. Virdis, S., Scarano, C., Cossu, F., Spanu, V., Spanu, C. and De Santis, E.P. 2010. Antibiotic resistance in Staphylococcus aureus and coagulase negative Staphylococci isolated from goats with subclinical mastitis. Vet. Med. Int. 2010, 517060. doi: 10.4061/2010/517060. Yahaghi, E., Imani, Fooladi, A.A., Amin, M., Mirnejad, R., Nezamzade, R. and Amani, J. 2014. Detection of Class I Integrons in Staphyloacoccus aureus isolated from clinical samples. Iran Red Crescent Med. J. 16, e16234. doi:10.5812/ircmj.16234. Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S. and Madden, T.L. 2012. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 13, 134. doi:10.1186/1471-2105-13-134. | ||
| How to Cite this Article |
| Pubmed Style Sarhan SR, Hashim HO, Shuhaib MBSA. The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates. Open Vet J. 2019; 9(4): 339-348. doi:10.4314/ovj.v9i4.12 Web Style Sarhan SR, Hashim HO, Shuhaib MBSA. The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates. https://www.openveterinaryjournal.com/?mno=50203 [Access: July 01, 2025]. doi:10.4314/ovj.v9i4.12 AMA (American Medical Association) Style Sarhan SR, Hashim HO, Shuhaib MBSA. The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates. Open Vet J. 2019; 9(4): 339-348. doi:10.4314/ovj.v9i4.12 Vancouver/ICMJE Style Sarhan SR, Hashim HO, Shuhaib MBSA. The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates. Open Vet J. (2019), [cited July 01, 2025]; 9(4): 339-348. doi:10.4314/ovj.v9i4.12 Harvard Style Sarhan, S. R., Hashim, . H. O. & Shuhaib, . M. B. S. A. (2019) The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates. Open Vet J, 9 (4), 339-348. doi:10.4314/ovj.v9i4.12 Turabian Style Sarhan, Sarhan R, Hyder O Hashim, and Mohammed Baqur S Al Shuhaib. 2019. The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates. Open Veterinary Journal, 9 (4), 339-348. doi:10.4314/ovj.v9i4.12 Chicago Style Sarhan, Sarhan R, Hyder O Hashim, and Mohammed Baqur S Al Shuhaib. "The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates." Open Veterinary Journal 9 (2019), 339-348. doi:10.4314/ovj.v9i4.12 MLA (The Modern Language Association) Style Sarhan, Sarhan R, Hyder O Hashim, and Mohammed Baqur S Al Shuhaib. "The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates." Open Veterinary Journal 9.4 (2019), 339-348. Print. doi:10.4314/ovj.v9i4.12 APA (American Psychological Association) Style Sarhan, S. R., Hashim, . H. O. & Shuhaib, . M. B. S. A. (2019) The Gly152Val mutation possibly confers resistance to beta-lactam antibiotics in ovine Staphylococcus aureus isolates. Open Veterinary Journal, 9 (4), 339-348. doi:10.4314/ovj.v9i4.12 |

