Open Veterinary Journal, (2021), Vol. 11(3): 447–457
Original Research
10.5455/OVJ.2021.v11.i3.18
HDAC2/3 inhibitor MI192 mitigates oligodendrocyte loss and reduces microglial activation upon injury: A potential role of epigenetics
Mohamed A. Al-Griw1 , Mansur E. Shmela2, Mohamed M. Elhensheri3 and Emad M. Bennour4
, Mansur E. Shmela2, Mohamed M. Elhensheri3 and Emad M. Bennour4
1Department of Histology and Genetics, Faculty of Medicine, University of Tripoli, Tripoli, Libya
2Department of Preventive Medicine, Genetics & Animal Breeding, Faculty of Veterinary Medicine, University of Tripoli, Tripoli, Libya
3Rostock University Medical Centre, Rostock, Germany
4Department of Internal Medicine, Faculty of Veterinary Medicine, University of Tripoli, Tripoli, Libya
*Corresponding Author: Emad M. Bennour. Department of Internal Medicine, Faculty of Veterinary Medicine, University of Tripoli, Tripoli, Libya. Email: e.bennour [at] uot.edu.ly
Submitted: 25/04/2021 Accepted: 04/08/2021 Published: 28/08/2021
© 2021 Open Veterinary Journal
This is an Open Access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives License (http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial re-use, distribution, and reproduction in any medium, provided the original work is properly cited, and is not altered, transformed, or built upon in any way.
Abstract
Background: During development, oligodendrocyte (OL) lineage cells are susceptible to injury, leading to life-long clinical neurodevelopmental deficits, which lack effective treatments. Drugs targeting epigenetic modifications that inhibit histone deacetylases (HDACs) protect from many clinical neurodegenerative disorders.
Aim: This study aimed to investigate the therapeutic potential of histone deacetylase 2/3 (HDAC2/3) inhibitor MI192 on white matter (WM) pathology in a model of neonatal rat brain injury.
Methods: Wistar rats (8.5-day-old, n=32) were used to generate brain tissues. The tissues were cultured and then randomly divided into four groups and treated as following: group I (sham); the tissues were cultured under normoxia, group II (vehicle); DMSO only, group III (injury, INJ); the tissues were exposed to 20 minutes oxygen-glucose deprivation (OGD) insult, and group IV (INJ + MI192); the tissues were subjected to the OGD insult and then treated with the MI192 inhibitor. On culture day 10, the tissues were fixed for biochemical and histological examinations.
Results: The results showed that inhibition of HDAC2/3 activity alleviated WM pathology. Specifically, MI192 treatment significantly reduced cell death, minimized apoptosis, and mitigates the loss of the MBP+ OLs and their precursors (NG2+ OPCs). Additionally, MI192 decreased the density of reactive microglia (OX−42+). These findings demonstrate that the inhibition of HDAC2/3 activity post-insult alleviates WM pathology through mechanism(s) including preserving OL lineage cells and suppressing microglial activation.
Conclusion: The findings of this study suggest that HDAC2/3 inhibition is a rational strategy to preserve WM or reverse its pathology upon newborn brain injury.
Keywords: Brain injury, Epigenetics, MI192, Microglia, Oligodendrocytes.
Introduction
Acetylation is a key post-translational protein modification that regulates crucial intracellular pathways (Baltan et al., 2011; Narita et al., 2019). Protein acetylation is described as the transfer of an acetyl moiety from acetyl coenzyme A to the ε-amino group of a lysine residue (VanDrisse and Escalante-Semerena, 2019). Lysine acetylation is a reversible process, where it occurs by the action of histone acetyltransferases (HATs) and is reversed by histone deacetylases (HDACs) nuclear enzymes (Narita et al., 2019). In this context, HATs and HDACs enzymes modulate histone acetylation states to either tilt or maintain the balance between gene activation and repression, respectively (Marmorstein and Zhou, 2014). The actions of HATs and HDACs enzymes are not limited only to the histone substrates, but they can also modify non-histone proteins, including transcription factors such as p53 and signal transduction mediators (Haberland et al., 2009; Baltan et al., 2011). Therefore, HDACs predictably modulate important cellular events, including the cell cycle, differentiation, and apoptosis (Marampon et al., 2019).
HDACs are widely expressed by both glial and neuronal cells of rat brains (Broide et al., 2007). HDACs are classified into four classes. Class I HDACs (1–3, and 8) are found within the cell nucleus, where they can deacetylate histones. Class II HDACs (4–7, 9, and 10) shuttle between the nucleus, the cytoplasm, and the histones. They also deacetylate cytoplasmic proteins such as the microtubules. Class III HDACs, also known as the sirtuins, couple deacetylation to nicotinamide adenine dinucleotide (NAD) + hydrolysis. The single member of class IV HDACs, HDAC11, has common features with both class I and II HDACs (Gregoretti et al., 2004).
Chromatin remodeling (states of histone acetylation and deacetylation) is critical for shaping the stage-specific differentiation processes during normal oligodendrocyte (OL) lineage development and function (Gregath and Lu, 2018). The histone acetylation and deacetylation states also play a role in controlling OL lineage cell specification, differentiation, myelination, and myelin repair/regeneration in a variety of neurodegenerative processes diseases (Cunliffe and Casaccia-Bonnefil, 2006; Gregath and Lu, 2018).
Many model systems were used to investigate the molecular etiology of white matter (WM) pathology (Marinova et al., 2009; Leng et al., 2010; Yan et al., 2013; Gerace et al., 2015; Salvador et al., 2015). Within the frame of the central nervous system (CNS) studies, several model systems were employed and utilized either whole animals for in vivo experimentation, isolated tissue models for ex-vivo experiments, or cell cultures, where specific cell types were isolated and maintained for investigation (Fern and Moller, 2000; Lorenzini et al., 2000; Salter and Fern, 2005; Al-Griw et al., 2017, 2021). Each of those model systems has its own asset. However, within the study of neonatal ischemia, there has been a tendency to employ either ex-vivo tissue models or experimental animal models (Fern and Moller, 2000; Lorenzini et al., 2000; Salter and Fern, 2005). This reflects the need to study the injury mechanism(s) in the context of the complex nature of CNS cellular matrix, where the injury to one cell type would affect other cellular components within that complex matrix (Fern, 1998; Stys, 2004; Tekkok et al., 2007; Matute, 2010; Al-Griw et al., 2017).
HDAC inhibitors (HDACis) are a group of heterogeneous agents that inhibit HDACs enzymes and promote post-translational acetylation of lysine residues within nuclear and cytoplasmic proteins. They then can change their activity and function (McClure et al., 2018). Particularly, HDAC inhibition can significantly affect the acetylation status of histone proteins within chromatin, resulting in an increased genes expression relevant to a protective role from a brain insult (Haberland et al., 2009; Baltan et al., 2011). Furthermore, inhibition of deacetylation could equally promote the acetylation of non-histone proteins, like transcription factors and signal transduction mediators, and determine their interaction, localization, and stability (Glozak et al., 2005; Dokmanovic et al., 2007; Narita et al., 2019).
In addition, HDACis are of significant interest in curing many cancers, as they enhance cancer cells death (Minucci and Pelicci, 2006; Marampon et al., 2019). They were also found to accelerate the differentiation of neural precursor cells (Siegenthaler et al., 2007) and preserve neuronal cells in the face of many insults both in vitro (Langley et al., 2008; Uo et al., 2009; McClure et al., 2018) and in vivo (Langley et al., 2009; Yuan et al., 2018). Several drugs with HDAC inhibitory activity have been used in many neurodegenerative conditions (Chuang et al., 2009; Baltan et al., 2011; Yuan et al., 2018).
Non-selective class I and II HDACis, including trichostatin-A (TSA), valproic acid (VPA), sodium phenylbutyrate, and suberoylanilide hydroxamic acid (SAHA), are neuroprotective against excitotoxicity. Both sodium phenylbutyrate and VPA were found to induce the up-regulation of pro-survival and anti-apoptotic genes using rat neurons in vitro (Marinova et al., 2009; Leng et al., 2010). SAHA has also been shown to be neuroprotective in a WM ischemic stroke model using mouse optic nerves, where SAHA preserved WM architecture, function as well as OL and axonal survivability (Baltan et al., 2011). In spite they exhibit beneficial properties, TSA, VPA, and SAHA lack specificity and are correlated with significant in vivo side effects (Armon et al., 1996; Durham et al., 2017; Merck, 2018). In order to reduce those side effects together with keeping the promising neuroprotective ability of HDAC inhibitors, a better strategy would implicate specifically suppressing HDACs.
Activation and/or over-activation of specific HDACs are correlated with many neurodegenerative diseases. HDAC2 levels in the spinal cord and the motor cortex are higher in patients with amyotrophic lateral sclerosis than controls (Janssen et al., 2010). In addition, HDAC3 activation and/or over-expression are also involved in several neurodegenerative diseases and causes neural cell death (Bardai and D’Mello, 2011). Furthermore, both HDAC2 and HDAC3 negatively modulated memory (McQuown et al., 2011), but inhibition of class I HDACs was found to enhance memory function in an animal model of Alzheimer’s disease (Kilgore et al., 2010).
Unlike the other class I HDAC, the activity of HDAC1 is neuroprotective in Huntington’s disease and stroke, and that inhibition of HDAC1 activity can lead to neurodegeneration (Bates et al., 2006; Kim et al., 2008). Furthermore, a recent study suggests that HDAC1 and HDAC2 selective inhibitors are therapeutically useful for targeting neuroinflammation in neurodegenerative disease by modulating the acetylation levels and function of the non-histone protein(s) (Durham et al., 2017). Taken together, a compound that selectively inhibits HDAC2 and HDAC3, such as HDAC2/3 inhibitor MI192, but not HDAC1, could be therapeutically better for treating brain injuries and diseases than the currently available non-selective general class I and II HDAC inhibitors.
In the present study, we investigated the efficacy of HDAC2/3 inhibitor MI192 in an ex vivo model of developing WM ischemia. As inflammation is an important representative of the underlying pathogenic events during neonatal brain injury, we analyzed the microglial cell response to MI192 application. We found that the application of HDAC2/3 inhibitor MI192 after the injury preserved WM cellular architecture, reduced neural cell loss, and minimized apoptosis. This protection was associated with a preservation of maturing OLs and a suppression of reactive microglia (OX−42+).
Materials and Methods
Animals and housing conditions
A total of 32 Wistar rats, aged 8.5 days and weighing 34.2 ± 2.6 g, were used in this study. The animals were housed and bred according to the standard conditions (a 12-hours light: 12-hours dark cycle and 26±2°C temperature) in the animal facility of the Faculty of Science, University of Tripoli. Rats had food and water ad libitum. Minimal discomfort and pain of animals were assured during experimentation.
Brain tissue generation
Methods were based on previously published protocols (Stoppini et al. , 1991; Kluge et al., 1998; Ghoumari et al., 2005; Al-Griw et al., 2021). Briefly, brains from Wistar rats were used to generate cerebral WM tissues. Pups were sacrificed under anesthesia, and the cerebrum was dissected and transversely sliced at a thickness of 300-μm on a vibratome (Leica, Germany). Eight to ten tissue slices from 6 littermates were carefully transferred onto humidified 1.0 μm pore size cell culture inserts (Millipore, Falcon, UK) and placed in a six-well plate (Falcon). Cultures were maintained in 1 ml of serum-based medium (50% minimum essential medium Eagle (MEME, Sigma), 25% HBSS (Hanks balanced salt solution, Invitrogen), 20% normal horse serum (Invitrogen), 4.6 mM (v/v) L-glutamine (Sigma), 21 mM (v/v) D-glucose (Fisher Scientific, UK), 1% penicillin/streptomycin solution (Invitrogen), 4.2 μM (v/v) L-ascorbic acid (Aldrich-Sigma, Germany) and 11 mM (v/v) NaHCO3 at pH 7.2–7.4. Cultures were incubated in a humidified aerobic incubator (5% CO2) at 37°C for 3 days.
White matter injury induction
WM injury induction was simulated by exposing 7-day-old cultures to oxygen-glucose deprivation (OGD) insult for 20 minutes. Briefly, the tissue slices were transferred into filter-sterilized, deoxygenated glucose-free culture medium for 20 minutes in an anaerobic airtight chamber with a mixture of 95% N2 and 5% CO2 gas flow and 37 ± 0.5°C temperature. After the OGD insult, the cultures were washed three times using a fresh oxygenated culture medium containing 5 mg/ml D-glucose, complemented with 2% B27 and then returned to their culture conditions at 37°C and normoxic atmosphere (5% CO2). Sham cultures (controls) were maintained for the same time under normoxic conditions. The cultures were incubated for 3 days before being fixed for analysis.
Experimental group design
The tissue sections were randomly allocated to one of the four groups. In group I (sham control), the tissues were cultured under normoxic conditions. In group II (vehicle control), only 0.1% dimethyl sulfoxide (DMSO) (Sigma-Aldrich, Germany) was added, while in group III (injury (INJ)), the cultured tissues were exposed to 20 minutes OGD insult in an atmospheric perfusion airtight chamber. In group IV (INJ + MI192), the cultured tissues were subjected to OGD insult and treated with 1 μM HDAC2/3 inhibitor MI192 (Tocris Bioscience, Germany). The MI192 dose was selected based on prior studies (Al-Griw and Wood, 2016; Durham et al., 2017). Appropriate control was performed at the corresponding time points with this blocker to test its cytotoxicity. On culture day 10, the tissues were fixed for biochemical and histological examination.
Cell death assay
For cell death evaluation, a Live/Dead Viability/Cytotoxicity kit (Molecular Probes, Invitrogen, Germany) was used following manufacturer’s instructions and as previously described (Al-Griw et al., 2014). In brief, the harvested tissues were further incubated in the presence of a combined solution containing 4 μM ethidium homodimer-AM (EthD−1, red) and 2 μM calcein-AM (Cal, green) at 37°C for 35 minutes. They were then fixed in 4% paraformaldehyde (PFA, Sigma, Germany) in phosphate-buffered saline (PBS, Oxoid, Germany) for 30 minutes at room temperature.
4,6-diamidino-2-phenylindole (DAPI) staining and cell scoring
Nuclear morphological alterations of apoptosis were analyzed by the incorporation of fluorescent dye DAPI (Vector Laboratories, UK) as previously described (Bossenmeyer-Pourié et al., 2000). Briefly, the sections were fixed with 4% PFA for 30 minutes. The sections were then stained at room temperature for 10 minutes with DPAI. Five visual fields from each tissue section were analyzed, giving a total of 30 measurements per condition. Immunoreactive cells were counted only if their nuclei were visible with DAPI staining in the specific section. Cell counts were expressed as the percentage of immunoreactive cells to the total number of nuclei present. In each experimental condition, the total number of nuclei was also compared to ensure that differences observed between the groups were not attributable to a change in the number of cells. DAPI-labelled pyknotic nuclei were counted in the same fields as immunoreactive cells.
Immunocytochemistry
For immunocytochemical analysis, the cultured tissues were fixed in 4% PFA in PBS for 60 minutes at room temperature. Following three washes with PBS, the tissues were mounted on glass and were then blocked for 1 hour at room temperature with a serum blocking solution (10% normal goat serum) (MP Biomedical, Germany), 0.25% Triton 100-X (Sigma, Germany) in PBS. The tissue sections were incubated with 100 μl of primary anti-myelin basic protein (MBP) antibodies (#PD004; MBL, Germany), anti-NG2 (AB5320; Millipore, Germany), and anti-OX-42 (GR15599-3; Abcam, Germany), diluted in PBS (1:300), for 90 minutes at room temperature or overnight in the dark at 4°C. The tissue sections were then washed three times (5 minutes each) in PBS, stained with secondary antibodies (Alexa Fluor® 488 and 633) (#A21070 and #A11008, Invitrogen, Molecular Probes, Germany), diluted in PBS (1:1,000), for 2 hours at room temperature in the dark, and then washed three times in PBS. To examine the specificity of immunolabelling with the antibodies, control tissues were processed without primary antibodies, which resulted in no immunostaining.
Microscopy
The tissue sections were scanned and imaged using confocal microscopy (Leica, Germany) for three areas of interest per tissue section. A total of 30 optical sections of 1μm thickness at 1,024 × 1,024 pixel size were collected in the z-axis from a single microscopic field using the 63X objective lens under fixed gain, laser power, pinhole, and photomultiplier tubes (PMT) settings.
To compare and quantify immunohistochemical staining, the sections were processed concurrently. Flattened image stacks were opened in ImageJ image analysis software (National Institute of Health (NIH), USA), thresholded with constant pixel intensity level settings to a white background, and then analyzed with the analyze particles function. All quantifications were performed independently by a blind observer.
Statistics
Findings were statistically analyzed using the GraphPad Prism software version 7.0. All data represent the mean ± SEM for at least four independent experiments performed in triplicate. Normality was assessed using the computerized Kolmogorov-Smirnov test. For data with normal distributions, single between-group comparisons were made with the student paired t-test. Multiple comparisons were made with one-way analysis of variance followed by a post-hoc test for multiple comparisons Dunnett’s multiple-comparisons tests to detect pairwise between-group differences. p-value < 0.05 was considered to be statistically significant.
Ethical approval
Ethical approval for animal experiments was obtained from the Research Ethics Committee at the Biotechnology Research Center, Tripoli, Libya (Reference BEC-BTRC 8-2019).
Results
HDAC2/3 inhibitor MI192 reduces neural cell death upon injury
To investigate the potential role of HDAC2/3 inhibitor MI192 on the integrity of WM elements upon injury, the cell death was measured using live/dead assay. The results demonstrated the presence of dead cells (EthD−1, red) and viable cells (Cal, green) in all experimental groups (Fig. 1A). Specifically, we found that the injury triggered a significant (p < 0.001) increase in the percentage of dead cells in the INJ group (63.15% ± 7.68%, F =20.96) compared to the controls (9.56% ± 2.98%) (Fig. 1A).
On the other hand, MI192 treatment significantly reduced the percentage of neural cell death compared to the INJ group (22.04% ±7.1% for MI192, p =0.0002) (Fig. 1B) and resulted in a level of cell death not significantly different from controls (p =0.39; data not shown). The vehicle alone (DMSO) had no effect when applied under normoxia (Fig. 1B).
HDAC2/3 inhibitor MI192 minimizes apoptosis upon injury
We next asked whether treatment with HDAC2/3 inhibitor MI192-induced protection after injury involved changes in apoptosis among WM cellular elements. To this end, DAPI-nuclei labeling was carried out to measure apoptosis in all experimental groups. Compared with the nuclei of the sham group, which exhibited a round or oval shape and were used as a control, insult exerted a marked impact on the induction of cell apoptosis, as distinct nuclear morphological changes of apoptosis were noticed, including chromatin condensation, fragmentation, and apoptotic body formation (Fig. 2A). Specifically, the apoptotic cells were significantly (F =4.432; p =0.022) more abundant in the INJ group compared to the controls, and the treatment with MI192 significantly reduced their density (p =0.04) (Fig. 2B).
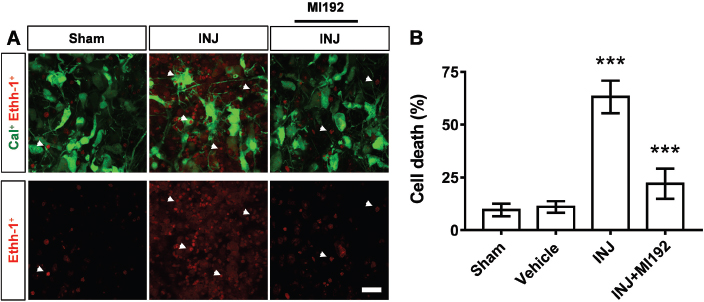
Fig. 1. (A and B) HDAC2/3 inhibitor MI192 reduced cell death after injury. Tissues were exposed to the conditions of sham, vehicle, INJ alone or with 1 μM MI192. (A): Immunofluorescent images of viable cells [calcein-AM (Cal), green] and nuclei of dead cells [ethidium homodimer-AM (EthD−1), red, white arrow heads]. Scale bar: 20 μm. (B): Quantification of cell death. Data are presented as mean ± SEM. (***) indicates p < 0.001.
HDAC2/3 inhibitor MI192 restores MBP+ OLs and NG2 + OPCs upon injury
The various cellular elements of neonatal WM are vulnerable to injury (Tekkok et al., 2007). The most susceptible WM cellular elements to damage are those of the OL lineage cells (Matute et al., 2007). Because the state of histone acetylation and deacetylation are critical for control of OL lineage cell specification, differentiation, myelination, and myelin repair/regeneration in a variety of neurodegenerative diseases (Cunliffe and Casaccia-Bonnefil, 2006; Gregath and Lu, 2018), we postulated here that inhibition of HDAC2/3 activity by MI192 might protect against OL lineage loss. To investigate that, we immunostained cultured tissues with an antibody to MBP, a marker of maturing OLs (Fig. 3A). The results showed that MBP+ OLs percent was significantly less abundant in the INJ group (7.38% ± 0.75%) compared to controls (18.21% ± 1.95%) (F =16.87, p < 0.001) (Fig. 3B). Interestingly, these negative effects were abolished by the addition of HDAC2/3 inhibitor MI192. Precisely, the MI192 treatment significantly enhanced the percent of MBP+ OLs (14.14% ± 1.75%) compared to that seen in the INJ group (p < 0.01) (Fig. 3B).
We then evaluated the impact of HDAC2/3 inhibitor MI192 on oligodendrocyte precursor cells (OPCs) after injury. For that, we immunostained cultured tissues with antibodies to NG2, an early OPC marker. The morphology of OPCs conformed to previous descriptions, is characterized by small, irregular, or rounded cell bodies with a few highly branched short processes (Levine and Card, 1987). The results revealed that NG2+ OPCs percentage were significantly less abundant ( F =10.02, p =0.0003) in the INJ group (5.63% ± 0.88%) compared to the controls (15.19% ± 1.34%) (Fig. 3C and D) and that treatment with MI192 significantly ( p =0.0129) enhanced the percentage of NG2+ OPCs (13.08% ± 1.02%) as compared to INJ group (Fig. 3D).
HDAC2/3 inhibitor MI192 suppresses microglial cell activation upon injury
Microglia are respondents in multiple forms of CNS insults (Miller et al., 2007). Activated microglia play a major role in losing or replacing OL lineage cells and myelin in normal and pathological conditions (Miller et al., 2007; Domingues et al., 2016; Traiffort et al., 2020). To explore microglial activation in this model, we quantified reactive microglia (OX−42+) in the immunolabeled cultured slices of cerebellar tissues. The results showed that the microglial marker OX−42 staining demonstrated the presence of OX−42+ microglia in all experimental groups (Fig. 4A). Specifically, it was found that OX−42+ microglia were more abundant (F =13.93, p < 0.001) in the INJ group (18.78% ± 1.47%) compared to the controls (6.0% ± 0.54%) (Fig. 4A). These findings correlate with OL lineage cell loss, as shown earlier (Fig. 3). On the other hand, MI192 treatment significantly ( p =0.0061) reduced the density of OX−42+ microglia cells (11.48% ± 1.93%) (Fig. 4B). Because MI192 suppresses reactive microglia in the INJ group, we reasoned that this might influence the survival of OLs. Taken together, these findings suggest that epigenetic manipulation after injury may have a role in mediating microglial cell activation.
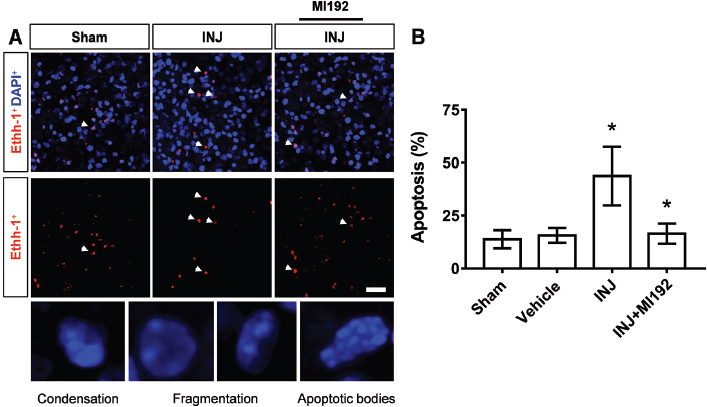
Fig. 2. (A and B) HDAC2/3 inhibitor MI192 minimized apoptotic cell death after injury. Tissues were exposed to the conditions of sham, vehicle, INJ alone or with 1 μM MI192. (A): Panoramic DAPI staining pattern in all groups (upper panel) and the morphological microscopic patterns of delayed nuclear condensation, fragmentation, and apoptotic bodies which have been used for judging the cell death profile in each group (lower panel). Scale bar: 20 μm. 200X magnification. (B): Quantification of cell apoptosis. Data are presented as mean ± SEM. (*) indicates p < 0.05.
Discussion
The current work demonstrated that the inhibition of HDAC activity, specifically HDAC2/3, after the OGD insult episode could preserve WM architecture and that this impact was mainly mediated by enhancement of cell survival, apoptosis reduction, inhibition of microglia reactivity, and preservation of maturing OLs MBP+ OLs and their precursors (NG2+ OPCs). Therefore, therapeutic potential for using HDAC inhibitors in preventing or reversing WM pathology could exist.
Preterm newborns show the greatest propensity for brain injury because of ischemic events, infection and/or inflammation, and a consequent remarkable WM injury, compared to newborns carried to the full term. Therefore, such causes represent major factors in the pathogenesis of prominent WM injury (Deng, 2010; Gilles et al., 2018). Based on HDACis ability to increase histone acetylation, alter the transcription of associated genes, and provide tissue protection, the potential of those inhibitors as therapeutic agents in a variety of diseases has emerged (Dinarello et al., 2011; McClure et al., 2018).
Models of cell cultures have been extensively used to examine the efficacy of a range of HDACis against WM injury (Marinova et al., 2009; Leng et al., 2010; Yan et al., 2013; Salvador et al., 2015). However, the ex vivo slice brain model system used in this study provides a more appropriate environment of the CNS with a mixture of cell types, an environment those homogeneous cell cultures do not offer. CNS neurodegeneration implicates a complex interplay between neurons and glia. Therefore, intact brain tissues maintain such a mix of different cell types and their interactions in a more complex multicellular environment.
In the current study, the brain tissue injury was induced by OGD insult followed by 3 days of reperfusion. Brain tissue OGD insult followed by the reperfusion period is an in vitro model that mimics the in vivo ischemia/reperfusion tissue injury (Shi et al., 2016). The underlying mechanism related to the reperfusion after transient OGD injury was attributed to the disruption of the membrane’s permeability and eventually leads to neuronal cell death (Gupta et al., 2013; Ureshino et al., 2014; Shi et al., 2016). Maintaining intracellular Ca2+ levels and activating Trk receptor tyrosine kinases have been used to preserve cells upon exposure to OGD followed by reperfusion (Gupta et al., 2013; Ureshino et al., 2014; Shi et al., 2016). Alterations in Ca2+ level increase cell vulnerability and oxidative stress in apoptosis progress (Ureshino et al., 2014). In addition, TrkB receptor tyrosine kinases, a family of transmembrane-receptor signaling systems, mediate pathways through downstream protein kinase C signaling to promote neuronal survival (Gupta et al., 2013). In the present study, we did not give information on the cell type most affected, the proportion of cell loss among glial or neuron cells, and whether an interaction occurred between glial cells and neurons. Nevertheless, HDAC inhibition prevented ischemia-mediated cell loss, suggesting that suppressing HDAC activity preserves all cell kinds vulnerable to ischemia-mediated death.
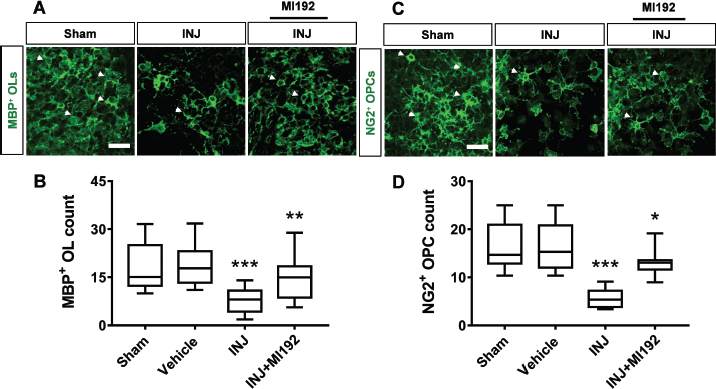
Fig. 3. (A–D) HDAC2/3 inhibitor MI192 restored MBP+ OL and NG2+ OPC loss after injury. Tissues were exposed to the conditions of sham, vehicle, INJ alone or with 1 μM MI192. (A): Immunofluorescent images of MBP+ OLs (white arrow heads, green) in all groups. Scale bar: 30 μm. (B): Quantification of MBP+ OLs. (C): Immunofluorescent images of NG2+ OPCs (white arrow heads, green). Scale bar: 30 μm. (D): Quantification of NG2+ OPCs. (OL): oligodendrocyte; (OPC): oligodendrocyte precursor cell. Data are presented as mean ± SEM. (*) indicates p < 0.05; (**) indicates p < 0.01; (***) indicates p < 0.001.
An attractive piece of the results of this study was the demonstrated efficacy of the HDAC2/3 inhibitor, MI192, when applied upon the OGD ischemic episode. Different pathways mediate injury to developing WM; for example, axons are injured by ionic degradation, whereas OLs and myelin are subject to excitotoxicity (Fern et al., 1995; Stys, 2004; Baltan et al., 2011) and the oxidative pathway damages WM elements because of free radical formation or by glutamate receptor activation (Oka et al., 1993; Baltan et al., 2011). The impact of general non-selective HDAC inhibitors in neuroprotection against glutamate excitotoxicity has been previously well documented in several reports (Ryu et al., 2005; Chen et al., 2006; Kazantsev and Thompson, 2008; Chuang et al., 2009; Kidd and Schneider, 2010, 2011).
The present study found that selective HDAC2/3 inhibitor MI192 robustly enhanced cell survival and reduced apoptosis in this ex vivo model. The exact underlying molecular mechanisms of protection by inhibition of HDAC2/3 are not fully understood yet. It was reported that administrated HDAC inhibitor sodium phenylbutyrate shows a concentration-dependent increase in neuronal cell survival (Ryu et al., 2005). Moreover, there was increased histone acetylation and prevention of apoptosis through an increase in the expression of Bcl−2 and a reduction in the release of cytochrome c from the mitochondria (Ryu et al., 2005). Therefore, this report could propose that the consistent protection via HDAC inhibition after the injury may be a result of the drug-related increasing in the transcription of pro-survival and anti-apoptotic genes during the development of WM injury. Still, that protection may also be mediated by targeting excitotoxic and oxidative pathways.
Another significant finding in this study was the demonstration that HDAC inhibition post-injury was associated with preserving OLs and their progenitors using cell-specific immunohistochemical labeling in this ex vivo model of neonatal WM pathology. Indeed, the importance of histone deacetylation for maturing OLs and immaturing OLs (OPCs) has been reported in previous experiments (Kouzarides, 2007; Kassis et al., 2014). Specifically, it was reported that that oligodendrogenesis after WM injury is correlated with distinct profiles of Class I (1–2) and Class II (4–5) HDACs in OL lineage cells (Kassis et al., 2014). This was explained by that the individual members of Class I and II HDACs could play divergent roles in OLs response to ischemia during WM remodeling (Kassis et al., 2014). Moreover, HDAC activity was shown to catalyze the removal of an acetyl group (deacetylation) from lysine residues of histone tails within nuclear chromatin and favors compaction of chromatin, leading to gene repression (Kouzarides, 2007).
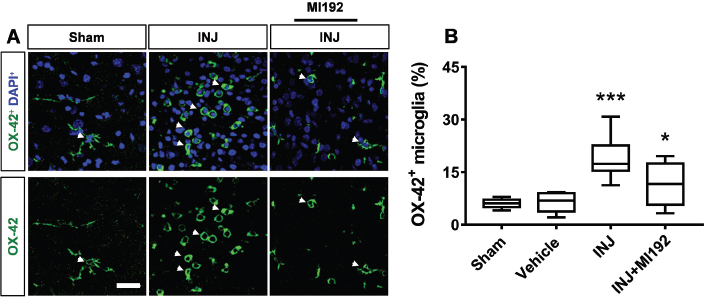
Fig. 4. (A and B) HDAC2/3 inhibitor MI192 suppressed microglial cell activation after injury. Tissues were exposed to the conditions of sham, vehicle, INJ alone or with 1 μM MI192. (A): Immunofluorescent images of OX−42+ cells (green) show an increased number of OX−42+ cells due to insult comparing to sham and INJ + MI192 groups. Blue color shows DAPI-stained nuclei. Scale bar: 30 μm. (B): Quantification of OX−42+ microglia. (*) indicates p < 0.05; (***) indicates p < 0.001.
Recent studies have primarily focused on using HDAC inhibitors, such as VPA, to promote WM recovery after stroke (Liu et al., 2012). These compounds target multiple classes of HDACs. The present study revealed that the insult promoted WM injury and that pharmacological inhibition of HDAC2/3 using the MI192 restored MBP+ OLs and NG2+ OPCs from injury. These findings suggest that selective HDAC inhibitors may provide more widespread protection and effectively treat various neurodegenerative disorders.
The results of this study also revealed a significant increase in the percentage of reactive OX−42+ microglia in response to injury. The treatment with HDAC2/3 inhibitor MI192 significantly reduced their density to the level observed under control conditions. This finding suggests that manipulation of epigenome after WM injury might have anti-inflammatory effects in this model, thereby changing the resultant microglia responses to injury. In this context, increasing evidence suggests that microglia recruitment is a hallmark of the inflammatory response to injury (Murugan et al., 2011; Patel et al., 2012; Cikla et al., 2016). Furthermore, activated microglia are critically involved in triggering WM injury (Deng, 2010). After injury, microglial cells are activated within minutes to hours (Morioka et al., 1991; Morin-Richaud et al., 1998). Microglia cells activation is associated with the production of different toxic molecules such as pro-inflammatory cytokines and reactive nitrogen and oxygen radicals (ROS) (Johnstone et al., 1999; Murphy, 2000), leading finally to the death of neural cells (Stavridis et al., 2005). It has also been reported that ROS and cytokines play a crucial role in OL death and delayed myelination (Merrill et al., 1993; Sherwin and Fern, 2005; Deng et al., 2008).
Conclusion
This work illustrates that selective HDAC2/3 inhibitor MI192 has significant protective ability in this ex vivo model of WM pathology. Although the precise mechanism(s) by which MI192 exerts its action was/were not elucidated, these findings imply that the protective role of MI192 maybe mediated by enhancing OL lineage cell population and suppressing microglial activation.
Acknowledgement
This work was funded by the University of Tripoli, Libya.
Conflict of interest
The authors declare that there is no conflict of interest.
Authors’ contribution
M. Al-Griw designed the study. M. Al-Griw, M. Shmela, M. Elhensheri, and E. Bennour contributed in interpreting the results and writing the manuscript.
References
Al-Griw, M.A., Alghazeer, R.O., Awayn, A., Shamlan, G., Eskandrani, A.A., Alnajeebi, A.M., Babteen, N.A. and Alansari, W.S. 2021. Selective adenosine A2A receptor inhibitor SCH58261 reduces oligodendrocyte loss upon brain injury in young rats. Saudi. J. Biol. Sci. 28, 310–316.
Al-Griw, M.A., Elnfati, A.S. and Ali, M.A. 2014. Gene delivery using adenoviral vector into the organotypic culture system of the chick retinae. J. Basic. Appl. Sci. 20, 28–36.
Al-Griw, M.A. and Wood, I.C. 2016. Histone deacetylase (HDAC) inhibition induces growth arrest and apoptosis in human medulloblastoma cells. Libyan J. Vet. Med. Sci. 1, 8–14.
Al-Griw, M.A., Wood, I.C. and Salter, M.G. 2017. Cerebellar organotypic slice culture system: a model of developing brain ischaemia. Life. Sci. J. 14, 89–98.
Armon, C., Shin, C., Miller, P., Carwile, S., Brown, E., Edinger, J.D. and Paul, R.G. 1996. Reversible parkinsonism and cognitive impairment with chronic valproate use. Neurology. 47, 626–635.
Baltan, S., Murphy, S.P., Danilov, C.A., Bachleda, A. and Morrison, R.S. 2011. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J. Neurosci. 31, 3990–3999.
Bardai, F.H. and D’Mello, S.R. 2011. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3beta. J. Neurosci. 31, 1746–1751.
Bates, E.A., Victor, M., Jones, A.K., Shi, Y. and Hart, A.C. 2006. Differential contributions of Caenorhabditis elegans histone deacetylases to huntingtin polyglutamine toxicity. J. Neurosci. 26, 2830–2838.
Bossenmeyer-Pourié, C., Koziel, V. and Daval, J. 2000. Effects of hypothermia on hypoxia-induced apoptosis in cultured neurons from developing rat forebrain: comparison with preconditioning. Pediatr. Res. 47, 385–391.
Broide, R.S., Redwine, J.F. and Aftahi, N. 2007. Distribution of histone deacetylases 1–11 in the rat brain. J. Mol. Neurosci. 31, 47–58.
Chen, P.S., Peng, G.S., Li, G., Yang, S., Wu, X., Wang, C.C., Wilson, B., Lu, R.B., Gean, P.W., Chuang, D.M. and Hong, J.S. 2006. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatry. 11, 1116–1125.
Chuang, D.M., Leng, Y., Marinova, Z., Kim, H.J. and Chiu, C.T. 2009. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends. Neurosci. 32, 591–601.
Cikla, U., Chanana, V., Kintner, D.B., Covert, L., Dewall, T., Waldman, A., Rowley, P., Cengiz P. and Ferrazzano, P. 2016. Suppression of microglia activation after hypoxia-ischemia results in age-dependent improvements in neurologic injury. J. Neuroimmunol. 291, 18–27.
Cunliffe, V.T. and Casaccia-Bonnefil, P. 2006. Histone deacetylase 1 is essential for oligodendrocyte specification in the zebrafish CNS. Mech. Dev. 123, 24–30.
Deng, W. 2010. Neurobiology of injury to the developing brain. Nat. Rev. Neurology. 6, 328–336.
Deng, Y., Lu, J., Sivakumar, V., Ling, E. and Kaur, C. 2008. Amoeboid microglia in the periventricular white matter induce oligodendrocyte damage through expression of proinflammatory cytokines via MAP kinase signaling pathway in hypoxic neonatal rats. Brain. Pathol. 18, 387–400.
Dinarello, C.A., Fossati, G. and Mascagni, P. 2011. Histone deacetylase inhibitors for treating a spectrum of diseases not related to cancer. Mol. Med. 17, 333–352.
Dokmanovic, M., Clarke, C. and Marks, P.A. 2007. Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer. Res. 5, 981–989.
Domingues, H.S., Portugal, C.C., Socodato, R. and Relvas, J.B. 2016. Oligodendrocyte, astrocyte, and microglia crosstalk in myelin development, damage, and repair. Cell. Front. Cell. Dev. Biol. 4, 71.
Durham, B.S., Grigg, R. and Wood, I.C. 2017. Inhibition of histone deacetylase 1 or 2 reduces induced cytokine expression in microglia through a protein synthesis independent mechanism. J. Neurochem. 143, 214–224.
Fern, R. 1998. Intracellular calcium and cell death during ischemia in neonatal rat white matter astrocytes in situ. J. Neurosci. 18, 7232–7243.
Fern, R. and Moller, T. 2000. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J. Neurosci. 20, 34–42.
Fern, R., Ransom, B.R. and Waxman, S.G. 1995. Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J. Neurophysiol. 74, 369–377.
Gerace, E., Landucci, E., Scartabelli, T., Moroni, F., Chiarugi, A. and Pellegrini-Giampietro, D.E. 2015. Interplay between histone acetylation/deacetylation and poly(ADP-ribosyl)ation in the development of ischemic tolerance in vitro. Neuropharmacology 92, 125–134.
Ghoumari, A.M., Baulieu, E.E. and Schumacher, M. 2005. Progesterone increases oligodendroglial cell proliferation in rat cerebellar slice cultures. Neuroscience 135, 47–58.
Gilles, F., Gressens, P., Dammann, O. and Leviton, A. 2018. Hypoxia-ischemia is not an antecedent of most preterm brain damage: the illusion of validity. Dev. Med. Child. Neurol. 60, 120–125.
Glozak, M., Sengupta, N., Zhang, X. and Seto, E. 2005. Acetylation and deacetylation of non-histone proteins. Gene. 19, 15–23.
Gregath, A. and Lu, Q.R. 2018. Epigenetic modifications-insight into oligodendrocyte lineage progression, regeneration, and disease. FEBS. Lett. 592, 1063–1078.
Gregoretti, I.V., Lee, Y.M. and Goodson, H.V. 2004. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J. Mol. Biol. 338, 17–31.
Gupta, V.K., You, Y., Gupta, V.B., Klistorner, A. and Graham, S.L. 2013. Trkb receptor signalling: implications in neurodegenerative, psychiatric and proliferative disorders. Int. J. Mol. Sci. 14, 10122–10142.
Haberland, M., Montgomery, R.L. and Olson, E.N. 2009. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10, 32–42.
Janssen, C., Schmalbach, S., Boeselt, S., Sarlette, A., Dengler, R. and Petri, S. 2010. Differential histone deacetylase mRNA expression patterns in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 69, 573–581.
Johnstone, M., Gearing, A.J. and Miller, K.M. 1999. A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J. Neuroimmunol. 93, 182–193.
Kassis, H., Chopp, M., Liu, X.S., Shehadah, A., Roberts, C. and Zhang, Z.G. 2014. Histone deacetylase expression in white matter oligodendrocytes after stroke. Neurochem. Int. 77, 17–23.
Kazantsev, A.G. and Thompson, L.M. 2008. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 7, 854–868.
Kidd, S.K. and Schneider, J.S. 2010. Protection of dopaminergic cells from MPP + -mediated toxicity by histone deacetylase inhibition. Brain. Res. 1354, 172–178.
Kidd, S.K. and Schneider, J.S. 2011. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 194, 189–194.
Kilgore, M., Miller, C.A., Fass, D.M., Hennig, K.M., Haggarty, S.J., Sweatt, J.D. and Rumbaugh, G. 2010. Inhibitors of class I histone deacetylase reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropyschopharmacology 35, 870–880.
Kim, D., Frank, C.L. and Dobbin, M.M. 2008. Deregulation of HDAC1 by p25/Cdk5 in Neurotoxicity. Neuron. 60, 803–817.
Kluge, A., Hailer, N.P., Horvath, T.L., Bechmann, I. and Nitsch, R. 1998. Tracing of the entorhinal-hippocampal pathway in vitro. Hippocampus. 8, 57–68.
Kouzarides, T. 2007. Chromatin modifications and their function. Cell. 128, 693–705.
Langley, B., Brochier, C. and Rivieccio, M.A. 2009. Targeting histone deacetylases as a multifaceted approach to treat the diverse outcomes of stroke. Stroke. 40, 2899–2905.
Langley, B., D’Annibale, M.A., Suh, K., Ayoub, I., Tolhurst, A., Bastan, B., Yang, L., Ko, B., Fisher, M., Cho, S., Beal, M.F. and Ratan, R.R. 2008. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21 waf1/cip1 in cell cycle-independent neuroprotection. J. Neurosci. 28, 163–176.
Leng, Y., Marinova, Z. and Reis-Fernandes, M.A. 2010. Potent neuroprotective effects of novel structural derivatives of valproic acid: potential roles of HDAC inhibition and HSP70 induction. Neurosci. Lett. 476, 127–132.
Levine, J.M. and Card, J.P. 1987. Light and electron microscopic localization of a cell surface antigen (NG2) in the rat cerebellum: association with smooth protoplasmic astrocytes. J. Neurosci. 7, 2711–2720.
Liu, X.S., Chopp, M., Kassis, H., Jia, L.F., Hozeska-Solgot, A., Zhang, R.L., Chen, C., Cui, Y.S. and Zhang, Z.G. 2012. Valproic acid increases white matter repair and neurogenesis after stroke. Neuroscience 3, 13–21.
Lorenzini, P., Pestalozza, S., Fortuna, S., Morelli, L., Volpe, M.T. and Meneguz, A. 2000. Metabotropic glutamate receptors and brain ischemia: differential effects on phospoinositide turnover in young and aged rats. Ann. Ist. Super. Sanita. 36, 453–457.
Marampon, F., Di-Nisio, V. and Pietrantoni, I. 2019. Pro-differentiating and radiosensitizing effects of inhibiting HDACs by PXD-101 (Belinostat) in in vitro and in vivo models of human rhabdomyosarcoma cell lines. Cancer. Lett. 461, 90–101.
Marinova, Z., Ren, M. and Wendland, J.R. 2009. Valproic acid induces function heat-shock protein 70 via Class I histone deacetylase inhibition in cortical neurons: a potential role of Sp1 acetylation. J. Neurochem. 111, 976–987.
Marmorstein, R. and Zhou, M.M. 2014. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold. Spring. Harb. Perspect. Biol. 6, a018762.
Matute, C. 2010. Calcium dyshomeostasis in white matter pathology. Cell. Calcium. 47, 150–157.
Matute, C., Alberdi, E., Domercq, M., Sanchez-Gomez, M., Perez-Samartin, A., Rodriguez-Antiguedad, A. and Perez-Cerda, F. 2007. Excitotoxic damage to white matter. J. Anat. 210, 693–702.
McClure, J.J., Li, X. and Chou, C.J. 2018. Advances and challenges of HDAC inhibitors in cancer therapeutics. Adv. Cancer. Res. 138, 183–211.
McQuown, S.C., Barrett, R.M., Matheos, D.P., Post, R.J., Rogge, G.A., Alenghat, T., Mullican, S.E., Jones. S., Rusche. J.R., Lazar, M.A. and Wood. M,A. 2011. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 31, 764–774.
Merck. 2018. Patient information ZOLINZA® (vorinostat) capsules. Darmstadt, Germany: Merch. Available via https://www.merck.com/product/usa/pi_circulars/z/zolinza/zolinza_ppi.pdf (Accessed January 2021).
Merrill, J.E., Ignarro, L.J., Sherman, M.P., Melinek, J. and Lane, T.E. 1993. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J. Immunol. 151, 2132–2141.
Miller, B.A., Crum, J.M. and Tovar, C.A. 2007. Developmental stage of oligodendrocytes determines their response to activated microglia in vitro. J. Neuroinflammation. 4, 28.
Minucci, S. and Pelicci, P.G. 2006. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer. 6, 38–51.
Morin-Richaud, C., Feldblum, S. and Privat, A. 1998. Astrocytes and oligodendrocytes reactions after a total section of the rat spinal cord. Brain. Res. 783, 85–101.
Morioka, T., Kalehua, A.N. and Streit, W.J. 1991. The microglial reaction in the rat dorsal hippocampus following transient forebrain ischemia. J. Cereb. Blood. Flow. Metab. 11, 966–973.
Murphy, S. 2000. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia 29, 1–13.
Murugan, M., Sivakumar, V., Lu, J., Ling, E.A. and Kaur, C. 2011. Expression of N-methyl D-aspartate receptor subunits in amoeboid microglia mediates production of nitric oxide via NF-kB signaling pathway and oligodendrocyte cell death in hypoxic postnatal rats. Glia 59, 521–539.
Narita, T., Weinert, B.T. and Choudhary, C. 2019. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 20, 156–174.
Oka, A., Belliveau, M.J., Rosenberg, P.A. and Volpe, J.J. 1993. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J. Neurosci. 13, 1441–1453.
Patel, H., Shaw, S.G., Shi-Wen, X., Abraham, D., Baker, D.M. and Tsui, J.C.S. 2012. Toll-Like receptors in ischaemia and its potential role in the pathophysiology of muscle damage in critical limb ischaemia. Cardiol. Res. Pract. 2012, 1–13.
Ryu, H., Smith, K., Camelo, S.I., Carreras, I., Lee, J., Iglesias, A.H., Dangond, F., Cormier, K.A., Cudkowicz, M.E., Brown R.H. and Ferrante, R.J. 2005. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J. Neurochem. 93, 1087–1098.
Salter, M.G. and Fern, R. 2005. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 438, 1167–1171.
Salvador, E., Burek, M. and Förster, C.Y. 2015. Stretch and/or oxygen glucose deprivation (OGD) in an in vitro traumatic brain injury (TBI) model induces calcium alteration and inflammatory cascade. Front. Cell. Neurosci. 9, 323.
Sherwin, C. and Fern, R. 2005. Acute lipopolysaccharide-mediated injury in neonatal white matter glia: role of TNF-a, IL-1B, and calcium. J. Immunol. 175, 155–161.
Shi, Z., Wu, D. and Yao, J.P. 2016. Protection against oxygen-glucose deprivation/reperfusion injury in cortical neurons by combining omega-3 polyunsaturated acid with lyciumbarbarum polysaccharide. Nutrients. 8, 41.
Siegenthaler, M.M., Tu, M.K. and Keirstead, H.S. 2007. The extent of myelin pathology differs following contusion and transection spinal cord injury. J. Neurochem. 24, 1631–1646.
Stavridis, S.I., Dehghani, F., Korf, H.W. and Hailer, N.P. 2005. Characterisation of transverse slice culture preparations of postnatal rat spinal cord: preservation of defined neuronal populations. Histochem. Cell. Biol. 123, 377–392.
Stoppini, L., Buchs, P.A. and Muller, D. 1991. A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods. 37, 173–182.
Stys, P.K. 2004. White matter injury mechanisms. Curr. Mol. Med. 2, 113–130.
Tekkok, S.B., Ye, Z. and Ransom, B.R. 2007. Excitotoxic mechanisms of ischemic injury in myelinated white matter. J. Cereb. Blood. Flow. Metab. 27, 1540–1552.
Traiffort, E., Kassoussi, A., Zahaf, A. and Laouarem, Y. 2020. Astrocytes and microglia as major players of myelin production in normal and pathological conditions. Front. Cell. Neurosci. 14, 79.
Uo, T., Veenstra, T.D. and Morrison, R.S. 2009. Histone deacetylase inhibitors prevent p53-dependent and p53-independent Bax-mediated neuronal apoptosis through two distinct mechanisms. J. Neurosci. 29, 2824–2832.
Ureshino, R.P., Rocha, K.K., Lopes, G.S., Bincoletto, C. and Smaili, S.S. 2014. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox. Signal. 21, 123–137.
VanDrisse, C.M. and Escalante-Semerena, J.C. 2019. Protein Acetylation in Bacteria. Annu. Rev. Microbiol. 8, 111–132.
Yan, W., Fang, Z., Yang, Q., Dong, H., Lu, Y., Lei, C. and Xiong, L. 2013. SirT1 mediates hyperbaric oxygen preconditioning-induced ischemic tolerance in rat brain. J. Cereb. Blood. Flow. Metab. 33, 396–406.
Yuan, H., Li, H., Yu, P., Fan, O., Zhang, X., Huang, H., Shen, J. and Zhou, W. 2018. Involvement of HDAC6 in ischaemia and reperfusion-induced rat retinal injury. BMC. Ophthalmol. 18, 300.






