




| Research Article | ||
Open Vet. J.. 2025; 15(2): 774-784 Open Veterinary Journal, (2025), Vol. 15(2): 774-784 Research Article In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamicMaged Fouda1*, Mohammad J. S. Mantargi2, Mousa O. Germoush1, Moustafa Sarhan3,4, Modather F. Husein5 and Mohamed M. Abdel-Daim2,61Biology Department, College of Science, Jouf University, Sakaka, Saudi Arabia 2Department of Pharmaceutical Sciences, Pharmacy Program, Batterjee Medical College, Jeddah, Saudi Arabia 3Department of Biomedical Sciences, College of Clinical Pharmacy, King Faisal University, Al Hofuf, Saudi Arabia 4Department of Zoology, Faculty of Science, Al-Azhar University, Assuit, Egypt 5Department of Chemistry, College of Science, Jouf University, Sakaka, Saudi Arabia 6Pharmacology Department, Faculty of Veterinary Medicine, Suez Canal University, Ismailia, Egypt *Corresponding Author: Maged Fouda. Biology Department, College of Science, Jouf University, Sakaka, Saudi Arabia. Email: mmfouda [at] ju.edu.sa Submitted: 02/11/2024 Accepted: 31/12/2024 Published: 28/02/2025 © 2025 Open Veterinary Journal
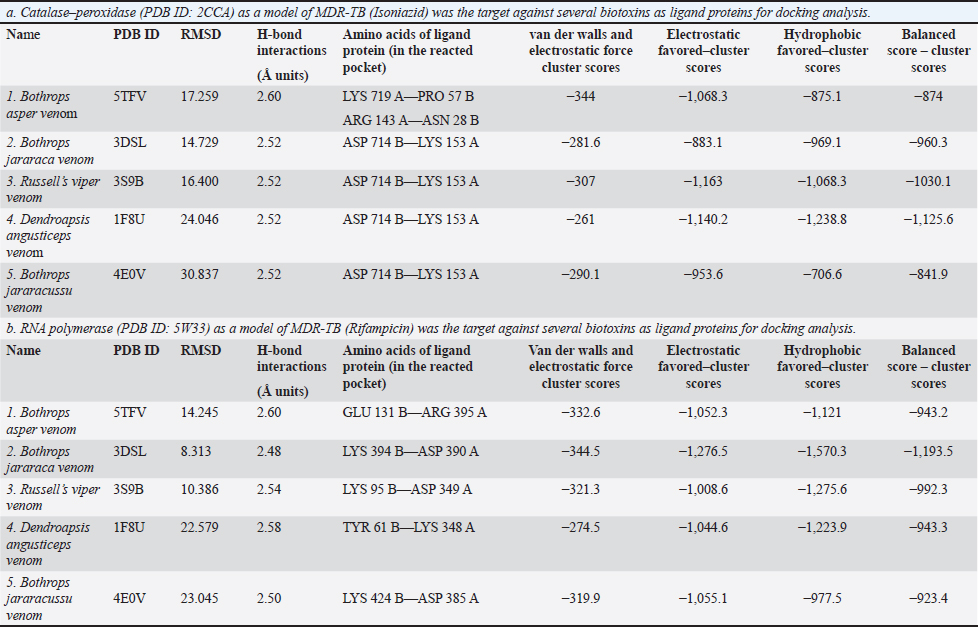
AbstractBackground: Mycobacterium tuberculosis (Mtb), a pathogen that belongs to the M. tuberculosis complex, causes tuberculosis (TB), an infectious bacterial disease. Although it usually affects the lungs and results in pulmonary TB, it can also lead to extra-pulmonary TB by affecting other regions of the body. TB, which ranks first on the list of infectious diseases that kill the most people, affects one-third of the world’s population and has a high mortality and morbidity rate. The clinical treatment of active TB infection mainly relies on the use of Isoniazid (INH) in combination with three other drugs—rifampin, pyrazinamide, and ethambutol. However, the situation is getting worse due to the rise of extensively drug-resistant tuberculosis (XDR-TB) and multidrug-resistant tuberculosis (MDR-TB). Finding more effective drugs is always a top priority. In this regard, animal venoms, such as snake toxins, contain antibacterial chemicals that have significant therapeutic properties and prevent bacterial infections and disease progression. This suggests that snake venom is a good natural source of promising novel anti-TB drugs. Aim: This study examines the snake venom protein’s capacity in silico to interrupt the intracellular enzymes of Mtb, which is responsible for the development of MDR-TB in humans. Methods: From Research Collaboratory for Structural Bioinformatics (RCSB)-Protein Data Bank, the active protein structure of catalase–peroxidase, RNA polymerase, and snake venom proteins was derived. Using molecular docking software such as PyRx, PyMOL, and Ligplot analyzers the interactions between those proteins and the targeted intracellular proteins were evaluated. Results: Our findings reveal fascinating affinities and interaction patterns between snake venom proteins and MDR-TB intracellular enzymes. Analysis of the effects of these interactions and their capacity to impair catalase–peroxidase and RNA polymerase showed that Russell’s viper venom proteins were active against the catalase–peroxidase system, whereas Bothrops jararaca venom proteins were active against the RNA polymerase system. Conclusion: This study highlights a prospective approach for advancing anti-TB agents by using snake venom proteins to inhibit the growth, replication, and transmission of MDR-TB. This will provide a basis for exploring pharmacophore-based drugs to combat MDR-TB infections. Keywords: MDR-TB, Catalase–peroxidase, RNA polymerase, Snake toxins, Molecular docking. IntroductionMycobacterium tuberculosis (Mtb) complex bacteria are the cause of tuberculosis (TB), an infectious disease that spreads through the air. TB is one of the most deadly infectious illnesses in humans. The first TB isolates were isolated in 1882 (Barberis et al., 2017). The current organism is said to have survived for 70,000 years and infected almost 2 million people worldwide (Doherty and Andersen, 2002). It has been deemed a public health emergency for the past 25 years because of its high rates of morbidity and mortality (Furin et al., 2019; Oliveira et al., 2021). It is estimated that 10.6 million incidences and 1.3 million deaths are reported annually because of TB worldwide in 2022 (WHO, 2024). Of these, rifampicin-resistant M. tuberculosis (RRMT) is of greater concern because it impacts human health to a major extent followed by isoniazid-resistant M. tuberculosis (IRMT). Low-to-middle-income nations, primarily in Africa and Southeast Asia, have higher rates of active TB illness, which is spread unevenly throughout the world (WHO, 2020). Despite all the efforts made to combat this illness, the growing number of Mtb strains that are resistant to drugs renders routine treatment ineffective and highlights the need for novel targeted therapeutic methods (Chan and Iseman, 2008; Kurz et al., 2016). Around 500,000 persons contracted Mtb strains resistant to rifampicin, the most potent first-line anti-mycobacterial medication, in 2019, according to the WHO. Seventy-eight percent of them were multidrug-resistant tuberculosis (MDR-TB), meaning that they were resistant to both isoniazid and rifampicin (Chan and Iseman, 2008; WHO, 2020). Furthermore, a more severe variant of MDR-TB known as extensively drug-resistant TB (XDR-TB) has surfaced. In addition to being resistant to fluoroquinolone and at least one of the three second-line injectables that are currently on the market (kanamycin, amikacin, and capreomycin), XDR-TB strains are also resistant to isoniazid and rifampicin. A total of 12,350 XDR-TB cases were reported in 2019 (Chan and Iseman, 2008, WHO, 2020). Antimicrobial resistance is therefore a major public health concern that must be addressed immediately. In this regard, antimicrobial peptides have emerged as viable options for the management of TB and other antimicrobial illnesses (Fox, 2013; Silva et al., 2016, Usmani et al., 2018). The earlier treatment history includes the development of para-amino salicylic acid in 1945 and its combination with streptomycin. in 1952, isoniazid in 1960, and ethambutol in 1970. A new chapter in TB treatment began with the addition of rifampicin. Nevertheless, this was quickly met by MDR-TB, which is resistant to drugs (Murray et al., 2015). Various reasons account for the failure of anti-TB drug therapy due to a lack of patient drug adherence, focus on drug susceptibility testing and its applications, and literacy and awareness of seriousness (Dooley et al., 2011). Therefore, to counter the challenge of MDR-TB, an additional molecule was added to the first-line regimen, including fluoroquinolones (Gureva et al., 2022) and macrolides (van der Paardt et al., 2015); however, certain resistance levels were observed (Prasad et al., 2018). For many centuries, medicinal plants and animal poisons have been used as alternative therapies for diseases, and they hold great promise for providing these needs. Despite their comparatively deadly contribution to global morbidity and mortality, venoms and toxins can be considered valuable therapeutic agents. Accordingly, substances with biological activity are also common in snake venom (Xie et al., 2003; Oguiura et al., 2023). Snake secretions are an important source of antimicrobials, and in silico studies can increase the success of searching for new molecules with therapeutic potential (Oguiura et al., 2023). Therefore, we conducted our study on snake venom screening for bioactive molecules against clinically isolated MDR-TB in order to meet the urgent need to identify high-efficacy medications against both drug-sensitive and drug-resistant Mtb strains with fewer side effects from some natural sources. Computational MethodsSelection of target and ligand proteinsThis study investigated the potential of snake venom proteins against Mtb enzymes. The 3D structures of catalase–peroxidase Protein Data Bank (PDB ID: 2CCA and resolution: 2.00 Å) and RNA polymerase (PDB ID: 5W33 and resolution: 2.85 Å) from MTb and five snake venom proteins (resolution range: 1.90–3.10 Å) were retrieved from the PDB (Berman et al., 2000; Goodsell et al., 2015). The details of the snake venom proteins used can be found in the following references: (Salvador et al., 2017) Bothrops asper venom—PDB ID: 5TFV and resolution: 2.54 Å; (Muniz et al., 2008) Bothrops jararaca venom—PDB ID: 3DSL and resolution: 2.70 Å; (Nakayama et al., 2011) Russell’s viper venom—PDB ID: 3S9B and resolution: 1.90 Å; (Kryger et al., 2000) Dendroaspis angusticeps venom—PDB ID: 1F8U and resolution: 2.90 Å; and (Ullah et al., 2012) B. jararacussu venom—PDB ID: 4E0V and resolution 3.10 Å. To prepare proteins for docking simulations, water molecules, ligands, and other nonprotein atoms (hetatms) were removed. Polar hydrogen was then added as necessary. Lipinski’s rule of five was applied to each snake venom protein to assess its potential as a therapeutic candidate. Protein–protein docking and analysisCluspro2 (Kozako et al., 2013, 2017; Vajda et al., 2017; Desta et al., 2020) was used for automated protein–protein docking simulations. PDB files of the MTb enzymes and snake venom proteins were submitted to the Cluspro2 server. The docking results included scores for various interaction parameters, such as the balanced cluster score, hydrophobic interactions, electrostatics, and van der Waals forces. The highest negative score was considered the lowest due to the data being represented in negative values. The resulting protein–protein interaction models were visualized and analyzed using PyMOL (Schrodinger, 2015) and LigPlot+ v2.2 (Laskowski and Swindells, 2011). These tools were used to identify interacting amino acids and analyze hydrogen bond lengths at the protein–protein interface. The study results, including binding affinity and interaction details, are presented in tables and graphs. Molecular dynamics simulationsThis study employed visual molecular dynamics (VMD) (Humphrey et al., 1996) and nanoscale molecular dynamics (NAMD) (Phillips et al., 2005) to investigate the effects of snake venom proteins on drug-resistant enzymes in Mtb. The enzymes catalase–peroxidase and RNA polymeras are targets of the commonly used anti-TB drugs isoniazid and rifampicin. The simulations were performed on a Windows 11 system equipped with an NVIDIA CUDA-enabled GPU to ensure efficient processing. VMD and NAMD were configured in the Windows 11 environment using Linux subsystems supported by Tcl and Python scripting languages. Commands were executed through the command prompt to generate log files, protein structure files (.psf), and trajectory data files (.dcd). The files were then loaded into VMD for further analysis of the simulation results (Stone et al., 2016; Phillips et al., 2020). ResultsThe global MDR-TB crisis can be solved with natural productsThe rapid spread of multidrug-resistant (MDR) Mtb since the mid-19th century has created a pressing global health crisis. This pathogen infects millions of people annually and remains a leading cause of death worldwide (Stephanie et al., 2021). To combat MDR-TB, developing novel drugs with potent enzyme-inhibiting properties and minimal adverse effects is essential. Computer-aided virtual screening has emerged as a valuable tool in drug discovery, accelerating the process and reducing costs (Laniado-Laborín, 2017). Molecular docking studies have identified potential lead compounds that bind to key MTb targets, such as the catalase–peroxidase system and RNA polymerase, which are critical for bacterial survival (DeVito and Morris, 2003). Natural products, including snake venom proteins from the Elapidae and Viperidae families (Mohamed Abd El-Aziz et al., 2019), represent promising sources of potential therapeutic compounds. Disrupting the activity of metabolically active enzymes involved in MTb growth and replication is a key strategy to combat this pathogen. Natural products, particularly those of microbial origin, offer a rich source of chemical diversity for drug discovery. Several natural product-based drugs, including antibacterial agents, have gained FDA approval (Andrei et al., 2019). Molecular docking and dynamic simulations of snake venom proteins against Mtb enzymesThis study conducted molecular docking simulations to evaluate the binding affinity of five snake venom proteins from the Viperidae, Elapidae, and Atractaspididae families against the Mtb enzymes catalase–peroxidase and RNA polymerase. The docking scores were used to rank the compounds based on their predicted binding affinity (Table 1a and b). The molecular docking analysis revealed strong interactions between the snake venom proteins and the MTb enzymes, with specific binding affinities observed for all five ligands. The model with the minimum docking score and highest binding affinity was considered the most stable interaction. PyMOL was used to visualize the protein–protein interactions (Agu et al., 2023), whereas Ligplot was employed to analyze amino acid interactions (Mishra and Dey, 2019). Hydrophobic and hydrogen bond interactions were identified between snake venom proteins and Mtb enzymes (Table 1a and b). Molecular dynamics simulations were performed on the top-scoring protein–protein complexes using the VMD/NAMD software suite from the Theoretical and Computational Biophysics Group at the National Institutes of Health. The simulation results, including root-mean-square deviation (RMSD), bond lengths, dihedral angles, and potential energy profiles, were analyzed. Molecular dynamics simulations reveal potential of snake venom proteins as anti-TB agentsMolecular dynamics simulations were conducted to evaluate the interactions between snake venom proteins and Mtb enzymes. Russell’s viper venom proteins exhibited strong binding affinity to catalase–peroxidase, with stable bond energies (~82,500 kcal/mol), reduced dihedral angles (18,800 to 18,200 kcal/mol), and minimal fluctuations in potential energy (~1,500 kcal/mol) and RMSD (<1.1 Å) (Table 2 and Fig. 1). Similarly, B. jararaca venom proteins showed favorable interactions with RNA polymerase, demonstrating stable bond energies (~35,000 kcal/mol), reduced dihedral angles (from 7,550 to 7,100 kcal/mol), and low potential energy fluctuations (~550 kcal/mol) and RMSD (<1.1 Å) (Table 2 and Fig. 2). These findings suggest that Russell’s viper and B. jararaca venom proteins are promising candidates for developing adjunctive therapies for RRMT and IRMT. Their ability to stabilize the MTb enzymes involved in drug metabolism could potentially enhance the efficacy of existing anti-TB therapies. Based on the table, all five venom proteins exhibited favorable interactions with catalase–peroxidase, as indicated by the relatively low RMSD values and negative cluster scores. The specific amino acid residues involved in these interactions vary among the venom proteins, suggesting different binding modes. Further analysis of these interactions could provide valuable insights into the mechanisms of action of these venom proteins. Lower RMSD values and higher negative scores for Van der walls, electrostatic, and hydrophobic interactions generally suggest stronger binding affinities. The amino acid residues involved in the interactions provide insights into the specificity of the venom protein for catalase–peroxidase. The relative contributions of van der Waals, electrostatic, and hydrophobic interactions can help identify the dominant forces driving the binding. Table 1. Protein–protein docking results of “Cluspro2,” analyzed through PyMOL, and visualized using Ligplot: (both a and b).
Table 2. Molecular dynamic simulations of Russell’s viper and B. jararaca venom proteins using catalase–peroxidase and RNA–polymerase, respectively.
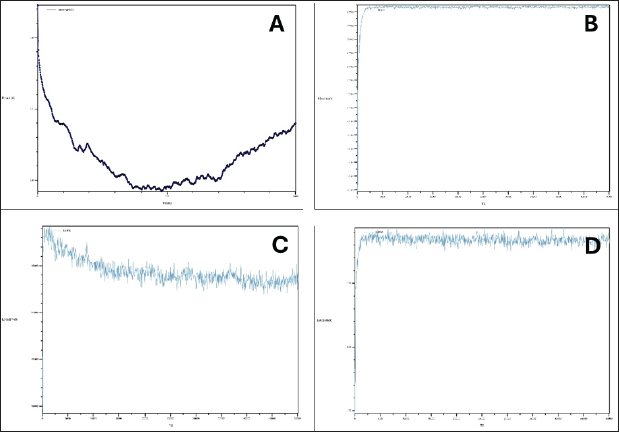
Fig. 1. Catalase–peroxidase interactions with Russell’s viper venom proteins. A: RMSD curve, B: Bonds curve, C: DIHED curve, and D: IMPRP curve.
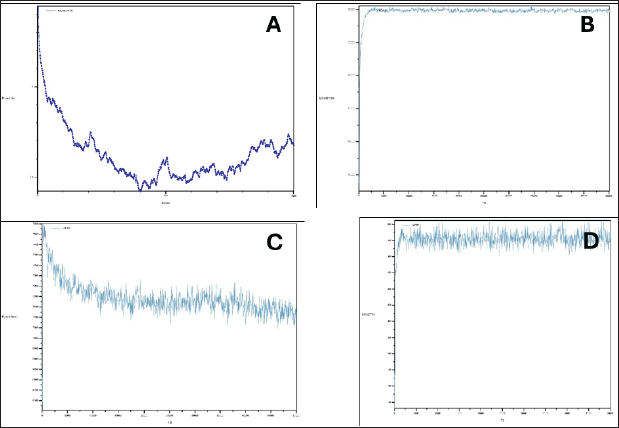
A: RMSD curve This plot shows the RMSD of the protein–protein complex over time. RMSD measures the average distance between the corresponding atoms in the protein–protein complex compared to a reference structure. From this figure, stable RMSD indicates that the protein–protein complex maintains a stable conformation throughout the simulation. A high RMSD indicates significant conformational changes or complex instability. B: Bond curve This plot likely represents the potential energy associated with bond stretching and bending within the protein–protein complex. A relatively stable bond energy curve suggests that the interactions between the proteins are stable and do not undergo significant changes during the simulation. C: DIHED curve This plot likely represents the potential energy associated with dihedral angle rotations in the protein–protein complex. Dihedral angles define the torsional twist around the bond. A stable DIHED curve indicates that the protein backbone conformation remained relatively unchanged during the simulation. D: IMPRP curve This plot likely represents the potential energy associated with improper torsions, which involve nonbonded interactions between atoms that are not directly connected. A stable IMPRP curve suggests that the overall geometry of the protein–protein complex remained consistent throughout the simulation. The relatively stable RMSD, bond, DIHED, and IMPRP curves suggest a stable and favorable interaction between catalase–peroxidase and Russell’s viper venom proteins. Further analysis of these plots could provide insights into the specific interactions and dynamics of the protein–protein complex. Fig. 2. RNA–polymerase interactions with B. jararaca venom proteins. A: RMSD curve, B: Bonds curve, C: DIHED curve, and D: IMPRP curve.
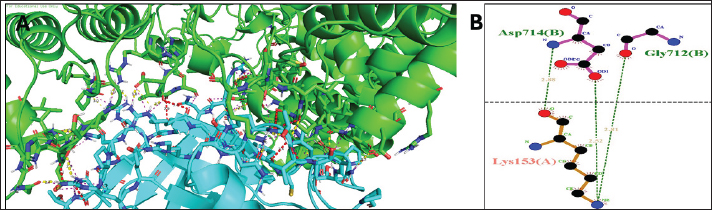
Details of MDR-TB catalase–peroxidase and RNA polymerase proteins and their interactions with various snake venom proteins of the family Elapidae and ViperidaeMolecular docking simulations were conducted to investigate the interactions between Mtb catalase–peroxidase and RNA polymerase with snake venom proteins from various species. Molecular interactions between MDR-TB catalase–peroxidase and snake venom proteinsMolecular docking simulations were conducted to investigate the interactions between Mtb catalase–peroxidase and snake venom proteins from B. asper and B. jararaca. The B. asper venom protein (PDB ID: 5TFV) exhibited strong binding affinity to MTb catalase–peroxidase, with favorable scores for van der Waals (−344), electrostatic (−1,068.3), hydrophobic (−875.1), and balanced (−874) interactions. The complex formed a hydrogen bond between lysine 719 A in catalase–peroxidase and proline 57 B in the venom protein (distance: 2.6 Å) (Table 1a). Similarly, B. jararaca venom protein (PDB ID: 3DSL) showed favorable interactions with MTb catalase–peroxidase, with scores of 281.6, 883.1, 969.1, and −960.3 for the respective interaction types. A hydrogen bond was formed between lysine 153 A in catalase–peroxidase and aspartic acid 714 B in the venom protein (distance: 2.52 Å) (Table 1a). These results suggest that B. asper and B. jararaca venom proteins have the potential to interact with MTb catalase–peroxidase and may be explored as potential therapeutic agents for TB. Russell’s viper venom (PDB ID: 3S9B)The complex exhibited strong binding affinity, with favorable scores for van der Waals (−307), electrostatic (−1,163), hydrophobic (−1,068.3), and balanced (−1,030.1) interactions. A hydrogen bond was formed between lysine 153 A in catalase–peroxidase and aspartic acid 714 B in the venom protein (distance: 2.52 Å) (Table 1a and Fig. 3A and B). Dendroapsis angusticeps venom (PDB ID: 1F8U)Similar to Russell’s viper venom, this complex displayed strong binding affinity, with scores of 261, 1,140.2, 1,238.8, and −1,125.6. A hydrogen bond was formed between lysine 153 A in catalase–peroxidase and aspartic acid 714 B in the venom protein (distance: 2.52 Å) (Table 1a). Bothrops jararacussu venom (PDB ID: 4E0V)The complex exhibited favorable interactions with scores of 290.1, 953.6, 706.6, and −841.9. A hydrogen bond was formed between lysine 153 A in catalase–peroxidase and aspartic acid 714 B in the venom protein (distance: 2.50 Å) (Table 1a). Molecular interactions between MDR-TB RNA polymerase and snake venom proteinsMolecular docking simulations were conducted to investigate the interactions between Mtb RNA polymerase and snake venom proteins from various species. Bothrops asper venom (PDB ID: 5TFV)The complex showed strong binding affinity, with scores of 332.6, 1,052.3, 1,121, and −943.2. A hydrogen bond was formed between arginine 395 A in RNA polymerase and glutamic acid 131 B in the venom protein (distance: 2.6 Å) (Table 1b). Bothrops jararaca venom (PDB ID: 3DSL)The complex exhibited favorable interactions with scores of 344.5, 1,276.5, 1,570.3, and −1,193.5. A hydrogen bond was formed between arginine 395 A in RNA polymerase and glutamic acid 131 B in the venom protein (distance: 2.48 Å) (Table 1b and Fig. 4A and B). Russell’s viper venom (PDB ID: 3S9B)The complex exhibited strong binding affinity, with favorable scores for van der Waals (−321.3), electrostatic (−1,008.6), hydrophobic (−1,275.6), and balanced (−992.3) interactions. A hydrogen bond was formed between aspartic acid 349 A in RNA polymerase and lysine 95 B in the venom protein (distance: 2.54 Å) (Table 1b). Dendroapsis angusticeps venom (PDB ID: 1F8U)The complex showed favorable interactions with scores of 274.5, 1,044.6, 1,223.9, and −943.3. A hydrogen bond was formed between lysine 348 A in RNA polymerase and tyrosine 61 B in the venom protein (distance: 2.58 Å) (Table 1b). Bothrops jararacussu venom (PDB ID: 4E0V)The complex exhibited strong binding affinity, with scores of 319.9, 1,055.1, 977.5, and −923.4. A hydrogen bond was formed between aspartic acid 385 A in RNA polymerase and lysine 424 B in the venom protein (distance: 2.50 Å) (Table 1b). DiscussionOvercoming the challenges posed by MDR Mtb requires innovative strategies to either strengthen the human immune response or directly target the bacteria’s metabolic and synthetic enzymes. While traditional anti-TB drugs like isoniazid act on the catalase–peroxidase enzyme complex and rifampicin acts on the DNA-dependent RNA polymerase (Tiberi et al., 2017) were initially effective, they are the first-line agents highly employed in the management of TB, reported to be at risk of failure with time as the bacteria is identified to have developed resistance against both the molecules by restructuring the protein expression hence termed as MDR-TB (Agusto et al., 2015). The emergence of MDR-TB has necessitated the search for novel therapeutic agents. This study investigated snake venom proteins belonging to the Elapidae and Viperidae families as potential inhibitors of MTb catalase–peroxidase and RNA polymerase, two essential enzymes for bacterial survival, as these families of snakes are a good source of untouched proteins for drug development against MDR-TB. (Nawarak et al., 2003; Salo-Ahen et al., 2021; Oliveira et al., 2022). Molecular docking and dynamic simulations revealed that venom proteins from Russell’s viper and B. jararaca exhibited strong binding affinity to these enzymes. Notably, Russell’s viper venom showed high affinity for catalase–peroxidase, which was identified to be resistant to the action of Isoniazid, while B. jararaca venom demonstrated strong binding to RNA polymerase, which was identified to be resistant to the action of Rifampicin (Table 1a and 1b) (Prasad et al., 2018), which are further established with the application of MD simulations (Durrant and McCammon, 2011). These interactions were characterized by stable protein–protein complexes and reduced enzyme activity, suggesting their potential to inhibit MTb growth. Fig. 3. (A and B): MDR-TB catalase–peroxidase (PDB ID 2CCA) with Russell’s viper venom (PDB ID 3S9B). The figure depicts the docked complex of Mtb catalase–peroxidase (PDB ID 2CCA) and Russell’s viper venom (PDB ID 3S9B). The colors may represent different protein chains or residues. The figure highlights a specific region of the complex in which the two proteins interact. The residues Asp714 B (from the venom protein) and Lys153A (from catalase–peroxidase) are shown in close proximity, suggesting a potential interaction. This figure provides a closer look at the interaction site identified in Figure 3A. The amino acid residues Asp714 B and Lys153A are described in detail, along with their side chains and potential hydrogen bonding interactions. The dashed lines likely represent hydrogen bonds between the two residues. These bonds contribute to the stability and specificity of the protein–protein complex. A and B: Together, these suggest a potential interaction between Mtb catalase–peroxidase and Russell’s viper venom. The specific interactions highlighted involve hydrogen bonds between Asp714B and Lys153A. This interaction may contribute to the binding affinity and specificity of the venom protein to the enzyme.
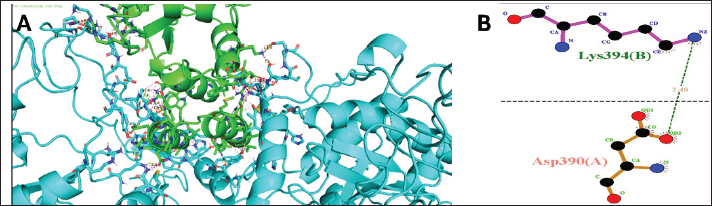
Fig. 4. (A and B): MDR-TB RNA polymerase (PDB ID 5W33) with B. jararaca venom (PDB ID 3DSL). MDR-TB RNA polymerase with snake venom protein (B. jararaca venom) (PDB ID: 3DSL) complex: molecular interactions with best Van der wall forces scores. Figure 4A depicts the docked complex of Mtb RNA polymerase (PDB ID 5W33) and B. jararaca venom (PDB ID 3DSL). The colors may represent different protein chains or residues. The figure highlights a specific region of the complex in which the two proteins interact. The residues Lys394B (from the venom protein) and Asp390A (from RNA polymerase) are shown in close proximity, suggesting a potential interaction. Figure 4B shows a closer look at the interaction site identified in Figure 4A. The amino acid residues Lys394B and Asp390A are shown in detail, along with their side chains and potential hydrogen bonding interactions. The dashed line likely represents a hydrogen bond between the two residues. This bond enhances the stability and specificity of the protein–protein complex. Figure 4A and B: together suggest a potential interaction between Mtb RNA polymerase and B. jararaca venom. The specific interactions highlighted involve hydrogen bonds between Lys394B and Asp390A. This interaction may contribute to the binding affinity and specificity of the venom protein to the enzyme.
While conventional antibiotics like fluoroquinolones have been used to treat MDR-TB, the development of bacterial resistance to these agents remains a significant concern, as the bacteria are slowly developing resistance to these agents (Hooper et al., 1987; El Sahly et al., 2011). The snake venom proteins of Russell’s viper and B. jararaca identified in this study offer a promising alternative for the development of novel anti-TB therapies. Interferons, being protein-based, are considered promising for treating viral infections, and the protein toxins under investigation could potentially serve as reliable sources for future anti-TB treatments (Mundra et al., 2023). Protein-based drugs have been successfully used to treat various diseases, including viral infections and critical disorders (Leader et al., 2008, Lagassé et al., 2017). The venom proteins investigated in this study could serve as a valuable source for developing new anti-TB agents. Given the increasing prevalence of MDR-TB, innovative therapeutic approaches are urgently required to address this global health threat. ConclusionThe global spread of MDR-TB presents a significant challenge to the scientific community. Developing novel drugs targeting key Mtb enzymes is a promising strategy to combat this disease. Computational methods, such as virtual screening, molecular docking, and in silico simulations, can accelerate drug discovery and reduce costs. This study investigated the potential of snake venom proteins as therapeutic agents against MDR-TB. Venom proteins from various snake species were screened against MTb using molecular docking and simulation techniques. Several compounds exhibited strong binding affinity and favorable interactions with the target proteins, suggesting their potential as anti-MDR-TB agents. The proteins have potential as anti-MDR-TB agents because they can inhibit the interaction between human cells and MDR-TB. The findings of this study highlight the potential of snake venom proteins as novel therapeutic candidates. Further research, including high-throughput screening, cell line studies, preclinical trials, and human clinical trials, is warranted to evaluate the efficacy and safety of these compounds for the treatment of MDR-TB. AcknowledgmentThe authors extend their appreciation to the Deanship of Scientific Research at Jouf University, Saudi Arabia, for funding this work through research grant number (DSR2021-03-03150). Conflicts of interestThe authors declare no conflicts of interest. FundingThis work was funded by the Deanship of Scientific Research at Jouf University under Grant Number (DSR2021-03-03150). Authors’ contributionsMMA is the designer of the research. MJ, MF, MS, and MFH analyzed the data and wrote the manuscript. MF, MMA, MS, and MOG revised the manuscript. MF applied for funding. All authors have read and approved the final manuscript. Data availabilityAll data generated or analyzed during this study are included in this article. ReferencesAgu, P.C., Afiukwa, C.A., Orji, O.U., Ezeh, E.M., Ofoke, I.H., Ogbu, C.O., Ugwuja, E.I. and Aja, P.M. 2023. Molecular docking as a tool for the discovery of molecular targets of nutraceuticals in diseases management. Sci. Rep. 13(1), 13398; doi:10.1038/s41598-023-40160-2 Agusto, F.B., Cook, J., Shelton, P.D. and Wickers, M.G. 2015. Mathematical model of MDR-TB and XDR-TB with isolation and lost to follow-up. Abstr. Appl. Anal. 2015, 828461; doi:10.1155/2015/828461 Andrei, S., Droc, G. and Stefan, G. 2019. FDA approved antibacterial drugs: 2018-2019. Discoveries 7(4), e102; doi:10.15190/d.2019.15 Barberis, I., Bragazzi, N.L., Galluzzo, L. and Martini, M. 2017. The history of tuberculosis: from the first historical records to the isolation of Koch’s bacillus. J. Prev. Med. Hyg. 58, E9–e12. Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H., Shindyalov, I.N. and Bourne, P.E. 2000. The protein data bank. Nucleic Acids Res. 28(1), 235–242; doi:10.1093/nar/28.1.235 Chan, E.D. and Iseman, M.D. 2008. Multidrug-resistant and extensively drug-resistant tuberculosis: a review. Curr. Opin. Infect. Dis. 21(6), 587–595; doi:10.1097/QCO.0b013 e328319bce6 Desta, I.T., Porter, K.A., Xia, B., Kozakov, D. and Vajda, S. 2020. Performance and its limits in rigid body protein-protein docking. Structure. 28(9), 1071–1081.e3; doi:10.1016/j.str.2020.06.006 DeVito, J.A. and Morris, S. 2003. Exploring the structure and function of the mycobacterial KatG protein using trans-dominant mutants. Antimicrob. Agents Chemother. 47(1), 188–195; doi:10.1128/AAC.47.1.188-195.2003 Doherty, T.M. and Andersen, P. 2002. Tuberculosis vaccine development. Curr. Opin. Pulm. Med. 8, 183–187. Dooley, K.E., Lahlou, O., Ghali, I., Knudsen, J., Elmessaoudi, M.D., Cherkaoui, I. and El Aouad, R. 2011. Risk factors for tuberculosis treatment failure, default, or relapse and outcomes of retreatment in Morocco. BMC Public Health. 11, 1–7; doi:10.1186/1471-2458-11-140 Durrant, J.D. and McCammon, J.A. 2011. Molecular dynamics simulations and drug discovery. BMC Biol. 9, 71; doi:10.1186/1741-7007-9-71 El Sahly, H.M., Teeter, L.D., Jost Jr, K.C., Dunbar, D., Lew, J. and Graviss, E.A. 2011. Incidence of moxifloxacin resistance in clinical Mycobacterium tuberculosis isolates in Houston, Texas. J. Clin. Microbiol. 49(8), 2942–2945; doi:10.1128/JCM.00231-11 Fox, J.L. 2013. Antimicrobial peptides stage a comeback: better understanding of the mechanisms of action, modification and synthesis of antimicrobial peptides is reigniting commercial development. Nat. Biotechnol. 31(5), 379–382; doi:10.1038/nbt.2572 Furin, J., Cox, H. and Pai, M. 2019. Tuberculosis. Lancet 393(10181), 1642–1656; doi:10.1016/S0140-6736(19)30308-3 Goodsell, D.S., Dutta, S., Zardecki, C., Voigt, M., Berman, H.M. and Burley, S.K. 2015. The RCSB PDB “Molecule of the Month”: inspiring a molecular view of biology. PLoS Biol. 13(5), e1002140; doi:10.1371/journal.pbio.1002140 Gureva, T., Turkova, A., Yablokova, E., Smirnova, P., Sveshnikova, O., Zolotaya, O., Nikishova, E., Heldal, E., Hinderaker, S., Seddon, J.A. and Mariandyshev, A. 2022. Fluoroquinolone preventive therapy for children exposed to MDR-TB. Int. J. Tuberc. Lung. Dis. 26(2), 171–173; doi:10.5588/ijtld.21.0443 Hooper, D.C., Wolfson, J.S., Ng, E.Y. and Swartz, M.N. 1987. Mechanisms of action of and resistance to ciprofloxacin. Am. J. Med. 82(4A), 12–20. Humphrey, W., Dalke, A. and Schulten, K. 1996. Visual molecular dynamics. J. Mol. Graph. 14(1), 33–8, 27–8; doi:10.1016/0263-7855(96)00018-5 Kozakov, D., Beglov, D., Bohnuud, T., Mottarella, S.E., Xia, B., Hall, D.R. and Vajda, S. 2013. How good is automated protein docking? Proteins: Struct. Funct. Bioinf. 81(12), 2159–2166; doi:10.1002/prot.24403 Kozakov, D., Hall, D.R., Xia, B., Porter, K.A., Padhorny, D., Yueh, C., Beglov, D. and Vajda, S. 2017. The ClusPro web server for protein–protein docking. Nat Protoc. 12(2), 255–278; doi:10.1038/nprot.2016.169 Kryger, G., Harel, M., Giles, K., Toker, L., Velan, B., Lazar, A., Kronman, C., Barak, D., Ariel, N., Shafferman, A., Silman, I. and Sussman, J.L. 2000. Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr D Biol Crystallogr. 56(Pt 11), 1385–1394; doi:10.1107/s0907444900010659 Kurz, S.G., Furin, J.J. and Bark, C.M. 2016. Drug-resistant tuberculosis: challenges and progress. Infect. Dis. Clin. N. Am. 30(2), 509–522; doi:10.1016/j.idc.2016. 02.010 Lagassé, H.A., Alexaki, A., Simhadri, V.L., Katagiri, N.H., Jankowski, W., Sauna, Z.E. and Kimchi-Sarfaty, C. 2017. Recent advances in (therapeutic protein) drug development. F1000Res 6, 113; doi:10.12688/f1000research. 9970.1 Laniado-Laborín, R. 2017. Clinical challenges in the era of multiple and extensively drug-resistant tuberculosis. Rev. Panam. Salud. Publica. 41, e167; doi:10.26633/RPSP.2017.167. Laskowski, R.A. and Swindells, M.B. 2011. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51(10), 2778–2786; doi:10.1021/ci200227u Leader, B., Baca, Q.J. and Golan, D.E. 2008. Protein therapeutics: a summary and pharmacological classification. Nat. Rev. Drug. Discov. 7(1), 21–39. doi:10.1038/nrd2399 Mishra, A. and Dey, S. 2019. Molecular docking studies of a cyclic octapeptide-cyclosaplin from sandalwood. Biomolecules 9(11), 740; doi:10.3390/biom 9110740 Mohamed Abd El-Aziz, T., Garcia Soares, A. and Stockand, J.D. 2019. Snake venoms in drug discovery: valuable therapeutic tools for life saving. Toxins (Basel) 11(10), 564; doi:10.3390/toxins11100564 Mundra, A., Yegiazaryan, A., Karsian, H., Alsaigh, D., Bonavida, V., Frame, M., May, N., Gargaloyan, A., Abnousian, A. and Venketaraman, V. 2023. Pathogenicity of type I interferons in Mycobacterium tuberculosis. Int. J. Mol. Sci. 24(4), 3919; doi:10.3390/ijms24043919 Muniz, J.R.C., Ambrosio, A.L.B., Selistre-de-Araujo, H.S., Cominetti, M.R., Moura-da-Silva, A.M., Oliva, G., Garratt, R.C. and Souza, D.H.F. 2008. The three-dimensional structure of bothropasin, the main hemorrhagic factor from Bothrops jararaca venom: insights for a new classification of snake venom metalloprotease subgroups. Toxicon 52(7), 807–816; doi:10.1016/j.toxicon.2008.08.021 Murray, J.F., Schraufnagel, D.E. and Hopewell, P.C. 2015. Treatment of tuberculosis. a historical perspective. Ann. Am. Thorac. Soc. 12, 1749–1759; doi:10.1513/AnnalsATS.201509-632PS Nakayama, D., Ben Ammar, Y., Miyata, T. and Takeda, S. 2011. Structural basis of coagulation factor V recognition for cleavage by RVV-V. FEBS Lett. 585(19), 3020–3025; doi:10.1016/j.febslet.2011.08.022 Nawarak, J., Sinchaikul, S., Wu, C.Y., Liau, M.Y., Phutrakul, S. and Chen, S.T. 2003. Proteomics of snake venoms from Elapidae and Viperidae families by multidimensional chromatographic methods. Electrophoresis. 24(16), 2838–2854; doi:10.1002/elps.200305552 Oguiura, N., Sanches, L., Duarte, P.V., Sulca-López, M.A. and Machini, M.T. 2023 Past, present, and future of naturally occurring antimicrobials related to snake venoms. Animals 13(4), 744; doi:10.3390/ani13040744 Oliveira, G.S., Costa, R.P., Gomes, P., Gomes, M.S., Silva, T. and Teixeira, C. 2021. Antimicrobial peptides as potential anti-tubercular leads: a concise review. Pharmaceuticals (Basel) 14(4), 323; doi:10.3390/ph14040323 Oliveira, A.L., Viegas, M.F., da Silva, S.L., Soares, A.M., Ramos, M.J. and Fernandes, P.A. 2022. The chemistry of snake venom and its medicinal potential. Nat. Rev. Chem. 6, 451–469; doi:10.1038/s41570-022-00393-7 Phillips, J.C., Braun, R., Wang, W., Gumbart, J., Tajkhorshid, E., Villa, E., Chipot, C., Skeel, R.D., Kalé, L. and Schulten, K. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26(16), 1781–1802; doi:10.1002/jcc.20289 Phillips, J.C., Hardy, D.J., Maia, J.D., Stone, J.E., Ribeiro, J.V., Bernardi, R.C., Buch, R., Fiorin, G., Hénin, J., Jiang, W., McGreevy, R., Melo, M.C.R., Radak, B.K., Skeel, R.D., Singharoy, A., Wang, Y., Roux, B., Aksimentiev, A., Luthey-Schulten, Z., Kalé, L.V., Schulten, K., Chipot, C. and Tajkhorshid, E. 2020. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153(4), 044130; doi:10.1063/5.0014475 Prasad, R., Gupta, N. and Banka, A. 2018. Multidrug-resistant tuberculosis/rifampicin-resistant tuberculosis: principles of management. Lung India 35(1), 78–81; doi:10.4103/lungindia.lungindia_98_17 Salo-Ahen, O.M.H., Alanko, I., Bhadane, R., Bonvin, A.M.J.J., Honorato, R.V., Hossain, S., Juffer, A.H., Kabedev, A., Lahtela-Kakkonen, M., Larsen, A.S., Lescrinier, E., Marimuthu, P., Mirza, M.U., Mustafa, G., Nunes-Alves, A., Pantsar, T., Saadabadi, A., Singaravelu, K. and Vanmeert, M. 2021. Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 9(1), 71; doi:10.3390/pr9010071 Salvador, G.H.M., dos Santos, J.I., Lomonte, B. and Fontes, M.R.M. 2017. Crystal structure of a phospholipase A2 from Bothrops asper venom: insights into a new putative “myotoxic cluster”. Biochimie 133, 95–102; doi:10.1016/j.biochi.2016.12.015 Schrodinger, L.L.C. 2015. The PyMOL molecular graphics system, Version 1.8. Silva, J.P., Appelberg, R. and Gama, F.M. 2016. Antimicrobial peptides as novel anti-tuberculosis therapeutics. Biotechnol. Adv. 34, 924–940; doi:10.1016/j.biotechadv.2016.05.007 Stephanie, F., Saragih, M. and Tambunan, U.S.F. 2021. Recent progress and challenges for drug-resistant tuberculosis treatment. Pharmaceutics 13(5), 592; doi:10.3390/pharmaceutics13050592 Stone, J.E., Hynninen, A.P., Phillips, J.C. and Schulten, K. 2016. Early experiences porting the NAMD and VMD molecular simulation and analysis software to GPU-accelerated openpower platforms. High Perform. Comput. 9945, 188–206; doi:10.1007/978-3-319-46079-6_14 Tiberi, S., Scardigli, A., Centis, R., D’Ambrosio, L., Muñoz-Torrico, M., Salazar-Lezama, M.Á., Spanevello, A., Visca, D., Zumla, A., Migliori, G.B. and Caminero Luna, J.A. 2017. Classifying new anti-tuberculosis drugs: rationale and future perspectives Int. J. Infect. Dis. 56, 181–184; doi:10.1016/j.ijid.2016.10.026 Ullah, A., Souza, T.A.C.B., Abrego, J.R.B., Betzel, C., Murakami, M.T. and Arni, R.K. 2012. Structural insights into selectivity and cofactor binding in snake venom l-amino acid oxidases. Biochem. Biophys. Res. Commun. 421(1), 124–128; doi:10.1016/j.bbrc.2012.03.129 Usmani, S.S., Kumar, R., Kumar, V., Singh, S. and Raghava, G.P. 2018. AntiTbPdb: a knowledgebase of anti-tubercular peptides. Database (Oxford) 2018, bay025; doi:10.1093/database/bay025 Vajda, S., Yueh, C., Beglov, D., Bohnuud, T., Mottarella, S.E., Xia, B., Hall, D.R. and Kozakov, D. 2017. New additions to the clusPro server motivated by CAPRI. Proteins 85(3), 435–444; doi:10.1002/prot.25219 van der Paardt, A.F., Wilffert, B., Akkerman, O.W., de Lange, W.C., van Soolingen, D., Sinha, B., van der Werf, T.S., Kosterink, J.G. and Alffenaar, J.W.C. 2015. Evaluation of macrolides for possible use against multidrug-resistant Mycobacterium tuberculosis. Eur. Respir. J. 46(2), 444–455; doi:10.1183/09031936.00147014 WHO. 2020. Global tuberculosis report 2020. Geneva, Switzerland: WHO. Available via https://www.who.int/ publications/i/item/9789240013131 (Accessed 28 January 2021) WHO. 2024. World tuberculosis day 2024, In World Tuberculosis Day 2024. Geneva, Switzerland: WHO, Vol. 2024. Available via https://www.who.int/campaigns/world-tb-day/2024. Vol. 2024, pp. Xie, J.P., Yue, J., Xiong, Y.L., Wang, W.Y., Yu, S.Q. and Wang, H.H. 2003. In vitro activities of small peptides from snake venom against clinical isolates of drug-resistant Mycobacterium tuberculosis. Int. J. Antimicrob. Agents. 22(2), 172–174; doi:10.1016/s0924-8579(03)00110-9 | ||
| How to Cite this Article |
| Pubmed Style Fouda M, Mantargi MJS, Germoush MO, Sarhan M, Husein MF, Abdel-daim MM. In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic. Open Vet. J.. 2025; 15(2): 774-784. doi:10.5455/OVJ.2025.v15.i2.26 Web Style Fouda M, Mantargi MJS, Germoush MO, Sarhan M, Husein MF, Abdel-daim MM. In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic. https://www.openveterinaryjournal.com/?mno=227123 [Access: June 27, 2026]. doi:10.5455/OVJ.2025.v15.i2.26 AMA (American Medical Association) Style Fouda M, Mantargi MJS, Germoush MO, Sarhan M, Husein MF, Abdel-daim MM. In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic. Open Vet. J.. 2025; 15(2): 774-784. doi:10.5455/OVJ.2025.v15.i2.26 Vancouver/ICMJE Style Fouda M, Mantargi MJS, Germoush MO, Sarhan M, Husein MF, Abdel-daim MM. In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic. Open Vet. J.. (2025), [cited June 27, 2026]; 15(2): 774-784. doi:10.5455/OVJ.2025.v15.i2.26 Harvard Style Fouda, M., Mantargi, . M. J. S., Germoush, . M. O., Sarhan, . M., Husein, . M. F. & Abdel-daim, . M. M. (2025) In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic. Open Vet. J., 15 (2), 774-784. doi:10.5455/OVJ.2025.v15.i2.26 Turabian Style Fouda, Maged, Mohammad J. S. Mantargi, Mousa O. Germoush, Moustafa Sarhan, Modather F. Husein, and Mohamed M. Abdel-daim. 2025. In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic. Open Veterinary Journal, 15 (2), 774-784. doi:10.5455/OVJ.2025.v15.i2.26 Chicago Style Fouda, Maged, Mohammad J. S. Mantargi, Mousa O. Germoush, Moustafa Sarhan, Modather F. Husein, and Mohamed M. Abdel-daim. "In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic." Open Veterinary Journal 15 (2025), 774-784. doi:10.5455/OVJ.2025.v15.i2.26 MLA (The Modern Language Association) Style Fouda, Maged, Mohammad J. S. Mantargi, Mousa O. Germoush, Moustafa Sarhan, Modather F. Husein, and Mohamed M. Abdel-daim. "In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic." Open Veterinary Journal 15.2 (2025), 774-784. Print. doi:10.5455/OVJ.2025.v15.i2.26 APA (American Psychological Association) Style Fouda, M., Mantargi, . M. J. S., Germoush, . M. O., Sarhan, . M., Husein, . M. F. & Abdel-daim, . M. M. (2025) In silico evaluation of snake venom proteins against multidrug-resistant Mycobacterium tuberculosis: A molecular dynamics study and simulation dynamic. Open Veterinary Journal, 15 (2), 774-784. doi:10.5455/OVJ.2025.v15.i2.26 |

