




| Review Article | ||
Open Vet. J.. 2025; 15(2): 594-600 Open Veterinary Journal, (2025), Vol. 15(2): 594-600 Review Article The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal modelNurina Titisari1*, Hafandi Ahmad2, Nurdiana Samsulrizal3, Ahmad Fauzi4 and Intan Shameha Abdul Razak21Department of Veterinary Physiology, Faculty of Veterinary Medicine, Universitas Brawijaya, Malang, Indonesia 2Department of Veterinary Preclinical Sciences, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Serdang, Malaysia 3Faculty of Applied Sciences, Universiti Teknologi MARA, Shah Alam, Malaysia 4Department of Veterinary Clinical Pathology, Faculty of Veterinary Medicine, Universitas Brawijaya, Malang, Indonesia *Corresponding Author: Nurina Titisari. Faculty of Veterinary Medicine, Universitas Brawijaya, Indonesia. Email: nurina_titisari [at] ub.ac.id Submitted: 02/12/2024 Accepted: 02/01/2025 Published: 28/02/2025 © 2025 Open Veterinary Journal
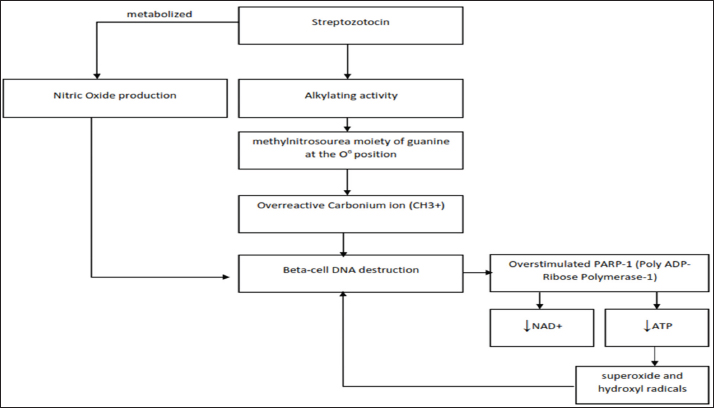
AbstractStreptozotocin (STZ) is a widely used chemical agent in biomedical research. It is primarily known for its ability to induce high blood glucose levels in animal models by selectively destroying pancreatic beta cells. Nonetheless, many studies have also used STZ to generate animal models of diabetic complications, such as Alzheimer’s disease (AD) animal models. STZ induction promotes hyperglycemia, which activates numerous mechanism pathways that result in the production of pathogenic AD characteristics, including beta-amyloid accumulation and neurofibrillary tangles. Numerous theories exist to elucidate the mechanisms underlying diabetes and AD; however, studies on the potential of an animal model of STZ-induced AD remain limited. Thus, this review summarizes the pathogenesis associated with STZ exposure, particularly in AD animal model studies related to diabetes. More specifically, this study will discuss the relationship between increased blood glucose levels after STZ injection and the process of beta-amyloid formation and insulin dysfunction in the brain. Keywords: Beta-amyloid, Brain, Hyperglycemia, Neuron, Tau protein. IntroductionAlzheimer’s disease (AD) is a neurodegenerative disorder characterized by memory loss, cognitive impairment, and behavioral changes. Neurofibrillary tangles (NFTs) made from hyperphosphorylated tau protein and senile plaques containing aggregated amyloid beta peptide (Aβ) are two main histopathological hallmarks of AD (Deture and Dickson, 2019). Furthermore, research into disease-modifying AD treatments is continuously ongoing. Currently, U.S Food and Drug Administration-approved AD treatments are limited to symptomatic therapies such as donepezil, galantamine, rivastigmine, and memantine (Yiannopoulou and Papageorgiou, 2020). For this purpose, experimental animal models that accurately replicate the developmental pathology of AD in humans are necessary. Transgenic mouse AD models are frequently used to identify the molecular pathways leading to memory deterioration. Previous research has shown that AD-transgenic animals are more accessible to generate NFTs than nontransgenic animals (Chen et al., 2013). Correspondingly, studies have shown that Aβ plaque development takes longer in regular animal models than in transgenic animals (Oliveira et al., 2021). However, there are limitations in using transgenic animals. According to Salkovic-Petrisic et al. (2013), transgenic AD models are not appropriate for investigating the origin, onset, and progression of pathological deposition of Aβ production in the brain under challenging circumstances that are not associated with mutations in the Aβ precursor protein (APP) gene. Indeed, high glucose increased Aβ production by inhibiting APP protein degradation rather than increasing APP gene transcription (Yang et al., 2013b). A new nontransgenic rat model has been proposed as a representative model of AD that uses streptozotocin (STZ) induction. STZ has a diabetogenic effect that induces irreversible pancreatic beta-cell damage through free radical generation and DNA damage (Ravelli et al., 2017). It is well known that the toxic effects of STZ dosage and its physiochemical properties continue to present major challenges for researchers. Factors such as preparation technique, solution stability, delivery route, and suitable dosage must be considered before establishing a study (Radenković et al., 2016). For instance, multiple low doses (MLDs) of STZ in rodents resulted in mild hyperglycemia similar to type 2 DM, whereas a single high dose of STZ resulted in severe hyperglycemia similar to type 1 DM (Ventura-Sobrevilla et al., 2011). Furthermore, studies have proven that STZ induction, either via intraperitoneal or intracerebroventricular induction, can induce neuroinflammation, leading to impaired brain insulin signaling and cognitive decline in experimental animals (Wang et al., 2019). Indeed, injecting STZ directly into the lateral ventricles did not affect blood glucose levels (Zhang et al., 2018), but it did alter the brain insulin signaling pathway, resulting in neuropathological and behavioral abnormalities (Grieb, 2016). Meanwhile, intraperitoneal injections of STZ may also compromise brain insulin signaling by attenuating phosphoinositide 3-kinase (PI3-K) signaling and increasing glycogen synthase kinase-3β (GSK-3β) activation, resulting in cognitive impairment in STZ-treated rats and possibly explaining early synaptic changes in sporadic AD (Ansari et al., 2023). In conclusion, both techniques may result in loss of cognition and an increase in Aβ deposits, total tau protein, and aggregated amyloid fragments in the brain. Therefore, this review discusses the relevant mechanism of STZ in generating animal models of AD due to hyperglycemia. The two basic mechanisms underlying diabetes and AD that are the subjects of this review are brain insulin resistance and amyloidogenesis (H. J. Lee et al., 2018). Characteristics of streptozotocin and its diabetogenic mechanismSTZ or -deoxy-2-([(methylnitrosoamino)carbonyl]amino)-D-glucopyranose] is a naturally occurring compound isolated from the soil bacteria Streptomyces achromogenes. This glucosamine-nitrosourea compound was originally developed as an anticancer agent for treating certain cancers of the islets of Langerhans and as an antibiotic for gram-negative bacteria (Abdollahi and Hosseini, 2014). Since 1963, It has been used in medical research to induce diabetes in experimental animals. STZ has a glucose analog structure with the addition of N-acetyl glycosamine and is toxic to pancreatic beta cells (Grieb, 2016). STZ penetrates the islet of Langerhans cells via glucose transporter 2 (GLUT2) in the plasma membrane and exerts pathological effects on the development of diabetes in animal models (Fig. 1). STZ spontaneously degrades upon entering cells to produce isocyanate and methyldiazohydroxide molecules. Isocyanate and methyldiazohydroxide compounds cause intramolecular carboxylation and alkylation of cellular components, respectively. The methyldiazohydroxide molecule disintegrates to form a highly reactive carbonium ion (CH3+) as the main key to deoxyribonucleic (DNA) alkylation (Goud et al., 2015). Next, the poly ADP-ribose polymerase-1 (PARP-1) enzyme becomes active when there is damage to the DNA chain. Overstimulating the PARP-1 enzyme depletes nicotinamide adenine dinucleotide + (NAD+), thereby reducing the amount of adenosine triphosphate (ATP) produced (Pieper et al., 1999). A lack of ATP impairs mitochondrial function, resulting in the inhibition of insulin synthesis and secretion. Ultimately, free radicals will develop, damaging pancreatic cells, and causing hyperglycemia (Eleazu et al., 2013). In addition, like other nitrosourea, STZ acts as a potential nitric oxide (NO) radical donor under in vivo conditions, mediating the destruction of pancreatic beta cells through DNA damage (Goud et al., 2015). Nitric oxide can increase guanyl silage activity and the formation of guanosine 3′,5′-cyclic monophosphate (cGMP), which produces reactive oxygen during cell damage. In conclusion, the release of NO, generation of reactive oxygen species (ROS), DNA alkylation, reduction in cell function, inhibition of glucosaminidase enzymes, reduction in insulin synthesis, and elevation of blood glucose levels are common effects of STZ (Šoltésová and Herichová, 2011). Streptozotocin alters insulin signaling in the brainBrain insulin resistance is defined as the incapability of brain cells to respond to insulin normally. According to a recent study, there is a direct link between brain insulin resistance and insulin deficiency in type 2 diabetes (De Sousa et al., 2020). However, both type 1 and type 2 diabetes mellitus share the commonality of insulin dysregulation, which is likely to have an impact on the brain (Talbot et al., 2012). Moreover, consuming a high-fat diet continuously can alter brain insulin signaling and cognitive dysfunction (Kothari et al., 2017). In the normal brain, insulin signaling activates PI3-K through phosphorylation at threonine 308 (Thr308), leading to protein kinase B or Ak strain transforming (AKT) kinase pathway activation. The activation of the AKT pathway will phosphorylate GSK-3β at serine 9 (Ser9) and inactivate it (Kleinridders et al., 2014). Meanwhile, it is well known that GSK-3β is a major tau kinase in the brain involved in the phosphorylation of tau at many hyperphosphorylation sites, including Ser199, Ser202, and Ser396 (Liu et al., 2007). Therefore, if insulin dysfunction occurs in diabetic disorders, it will block the AKT pathway and activate GSK-3β which then promotes tau hyperphosphorylation and NFTs (Burillo et al., 2021). Moreover, GSK-3β activation could potentially increase levels of Aβ and Aβ deposits in AD brains. Aβ accumulation can also induce tau protein phosphorylation and inactivate insulin receptor substrate-1 (IRS-1) substrate via c-Jun N-Terminal (JNT) Kinase signaling, disrupting insulin signaling (Ma et al., 2009). Intracerebroventricular injection of STZ causes central insulin resistance (IR) by disrupting IR signaling, decreasing expression of IR substrate type 1 (IRS-1), and increasing desensitization of IRs (Lester-Coll et al., 2006). Furthermore, STZ injection via ICV, besides interfering with insulin signaling, can also increase GSK-3β activation and tau phosphorylation. Researchers have suspected that brain insulin resistance is closely related to decreased PI3K-AKT signaling activity and overactivation of GSK-3β (Deng et al., 2009). On the other hand, IR using the intraperitoneal STZ injection method has also been widely reported by investigators (Yang et al., 2013a). Grieb (2016) stated that peripheral administration of STZ does not directly harm IR signaling, but secondary causes, such as insulin deficiency leading to high blood glucose levels. Bathina et al. (2017) provided evidence to support this claim, stating that there was a correlation between elevated blood sugar levels and lower protein expression of pAKT/AKT, pmTOR/mTOR, pPI3K/PI3K, Foxo1 as a secondary messenger of insulin signaling and also GSK-3β in STZ intraperitoneal injection of male Wistar rats. The results indicate that STZ causes insulin signaling dysregulation via the PI3K pathway.
Figure 1. Streptozotocin pathomechanism in pancreatic beta cells. The DNA methylating activity of the methylnitrosourea moiety of STZ, particularly at the O6 position of guanine, causes DNA damage and necrosis in pancreatic beta cells. When the DNA of the cell is damaged, PARP-1 is activated, which causes a decrease in ATP and NAD+, resulting in the formation of free radicals and damaging pancreatic cells. NO, which is generated following the metabolism of STZ, is another potential pathway that has also been linked to cell death. Streptozotocin-induced amyloid-β (Aβ) plaques and neurofibrillary tanglesIt is well-established that STZ causes hyperglycemia and senile plaque formation. In a histological study, senile plaques, also known as Aβ plaque, consist of Aβ protein, degenerative neuronal processes, and reactive nonneuronal cells (Drummond and Wisniewski, 2017). In fact, Aβ plaques are extracellular structures consisting of a central core and a corona. The central core in the middle of the structure is composed of extracellular beta-pleated amyloid aggregation, which constructs the Aβ peptide. Meanwhile, the corona, which encircles the central core, comprises degenerating neurons (primarily axons) containing tau protein and ubiquitin (Serrano-Pozo et al., 2011). Furthermore, Aβ peptide is a normal product of cellular metabolism generated from the proteolytic cleavage of a larger glycoprotein known as APP. Typically, APP is processed by enzymatic digestion α- and γ- secretase to form harmless peptide fragments in the non-amyloidogenic pathway, resulting in soluble Aβ ( Chen et al., 2017). This soluble protein can be transported from the brain to blood via the blood-brain barrier (BBB) via low-density lipoprotein receptor-related protein 1 (LRP1) to prevent Aβ peptide accumulation in the brain (Ramanathan et al., 2015). Moreover, hyperglycemia due to insulin deficiency can increase APP levels by reducing APP degradation and enhancing Aβ production (Yang et al., 2013b). Moreover, APP can lead to mitochondrial dysfunction and initiate stress signaling pathways (Reddy and Beal, 2008). In the diabetic brain, APP is processed via the amyloidogenic pathway. Amyloid plaques occur due to the accumulation of extracellular Aβ, derived from the enzymatic cleavage of APP through the β- and ɣ-secretase cleavage, which produces a 37–49 amino acid residue peptide. It then forms insoluble fibrous aggregates, causing amyloidosis and neurodegeneration (Murphy and Levine, 2010). In addition, two major isoforms of amyloid deposited in the AD brain are the 42-residue (Aβ42) as the main component and the 40-residue (Aβ40). Increased levels of Aβ42 or an increased ratio of Aβ42 induces Aβ fibril formations, and fibril accumulation develops into amyloid plaques, causing neurotoxicity through several mechanisms, such as the accumulation of free radicals, dysregulation of calcium homeostasis, inflammatory response, and activation of several signaling pathways pathway (Rajmohan and Reddy, 2017). Inflammatory cytokines such as interferon-γ (IFN-γ) induced microglial activation, which ultimately increases the production of inflammatory cytokines such as tumor necrosis factor-α (TNF-α). Furthermore, TNF-α will increase ROS levels and result in neuronal apoptosis. As brain-resident macrophages, microglia represent a therapeutic approach to coping with neuroinflammation (Sevenich, 2018). Most amyloid hypothesis in AD studies postulates that overproduction of Aβ peptide, or failure to clear this peptide, causes amyloid deposition and accumulation, which is thought to be involved in the formation of NFTs leading to neuronal loss (Ma et al., 2009). In 1963, previous research reported that the components of NFTs are two intertwined abnormal filaments called paired helical filaments (PHFs), between 15 and 32 mm in width, with a periodicity of around 80 nm. NFTs are formed by self-aggregates of hyperphosphorylated tau proteins. Tau protein was first discovered as a microtubule-associated protein (MAP) that stimulates tubulin assembly into microtubules, which localize primarily in the axon (Picone et al., 2020). Under normal physiological conditions, tau protein stabilizes neuronal microtubules. However, in AD pathology, tau hyperphosphorylation causes microtubule disassembly, resulting in the generation of aberrant aggregates that are toxic to neurons, leading to synaptic dysfunction and cell death. This mechanism has been observed in several neurological conditions collectively referred to as tauopathies (Orr et al., 2017). Evidence from a previous animal study reveals that two weeks after ICV administration, STZ injection was potentially responsible for Aβ and neurofilament protein expression and NFTs. ICV-STZ injection promoted Aβ deposition in the brain, increasing the expression of p-tau protein and cleaved caspase-3, whereas p-PI3K, p-AKT, and p-GSK-3β protein expression was inhibited (Abdallah et al., 2021). Another ICV STZ study revealed decreased IR mRNA expression and tau hyperphosphorylation at Ser396 predominantly in the hippocampus region (Gupta et al., 2018). Previous studies have demonstrated that tau phosphorylation at Ser396 and Ser404 is crucial for impaired microtubule assembly, both of which are primarily responsible for tau-mediated tubulin polymerization functional loss (Evans et al., 2000).
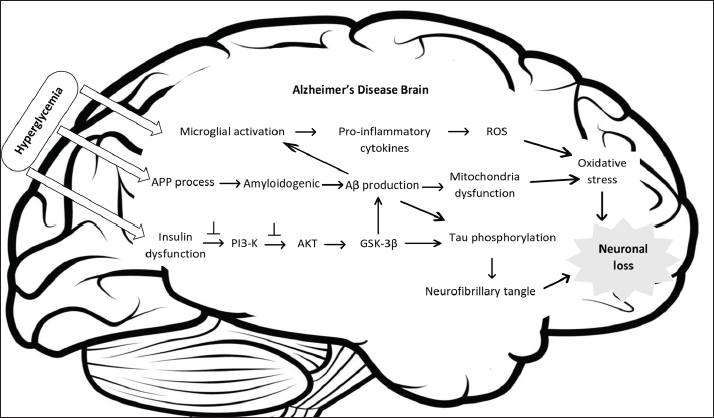
Figure 2. Summary of the impact of hyperglycemia after STZ injection in the animal brain. Hyperglycemia can affect macrophages and microglia as part of the innate immune system, increasing oxidative stress and inflammation, promoting amyloidogenic APP processing, and interrupting insulin receptors, causing insulin dysfunction. On the other hand, STZ injection via intraperitoneal elicited a different mechanism. Intraperitoneal injections of STZ significantly increase brain Aβ-42, β-secretase, and phosphorylated tau protein (Ali and Ali, 2022). Systemic injection of STZ cannot penetrate the BBB because of the lack of the STZ transporter GLUT2. Therefore, this method may influence brain metabolism indirectly because of high blood glucose levels, insulin deficiency, or other substances that may be toxic to the brain (Park, 2011). Systemic insulin deficiency in STZ-diabetic mice promotes reduced insulin-signaling pathway activity and increased GSK-3β activity in the brain (Jolivalt et al., 2010). GSK-3β has been suggested to play a role in APP processing leading to Aβ formation (Lee et al., 2003) and increased tau phosphorylation (Jope and Johnson, 2004). ConclusionBased on the presented discussion, Figure 2 shows the underlying mechanism of elevated blood glucose response following STZ injection. In addition, a few points that are worth noting in more detail are summarized below. First, STZ injection into the lateral ventricle of adult mice can impair glucose homeostasis and insulin signaling in the brain but not in the blood glucose body. Therefore, this method is not suitable for diabetes research to determine brain complications due to prolonged hyperglycemia. Second, the STZ effect discussed in this study is a general STZ effect, whereas different injection techniques exhibit distinct mechanisms. A single high dose directly damages pancreatic beta cells because of their cytotoxicity, causing extensive necrosis. In addition, MLDs induce limited apoptosis of pancreatic beta-cells, attracting mononuclear cells to eliminate the remaining cells (Cardinal et al., 2001). According to Grieb (2016), a single high dose has been widely used as a short-term diabetic rodent model, while multiple low-dose models have been proposed as long-term diabetic rodent models. Meanwhile, long-term hyperglycemia is the primary physiopathology of diabetic encephalopathy (Tomás Díaz-Gerevini et al., 2019). It is recommended to compare the mechanisms of each STZ approach in detail to construct an animal model of AD to aid investigators in conducting better investigations. Finally, the current study only examined two components contributing to hyperglycemia-induced AD. As a result, more research into other possible mechanisms should be undertaken to ensure comprehensive results. AcknowledgmentsNone. Conflict of interestThe authors declare that they have no conflicts of interest related to the publication of this manuscript. FundingNone. Authors’ contributionsNT: conceptualization and writing of the original draft. HA: conceptualization, review, and editing. AF: review and editing. NS and ISAR: supervision. All authors have read, reviewed, and approved the final manuscript. ReferencesAbdallah, H.M., El Sayed, N.S., Sirwi, A., Ibrahim, S.R.M., Mohamed, G.A. and Abdel Rasheed, N.O. 2021. Mangostanaxanthone IV ameliorates streptozotocin-induced neuro-inflammation, amyloid deposition, and tau hyperphosphorylation via modulating PI3K/Akt/GSK-3β pathway. Biology 10(12), 1298. Abdollahi, M. and Hosseini, A. 2014. Streptozotocin. In Encyclopedia of Toxicology, 3rd edition. Ed., Wexler, P. Oxford: Academic Press, pp: 402–404. Ali, S.K. and Ali, R.H. 2022. Effects of antidiabetic agents on Alzheimer’s disease biomarkers in experimentally induced hyperglycemic rat model by streptozocin. PLoS One 17(7), e0271138. Ansari, M.A., Al-Jarallah, A. and Babiker, F.A. 2023. Impaired insulin signaling alters mediators of hippocampal synaptic dynamics/plasticity: a possible mechanism of hyperglycemia-induced cognitive impairment. Cells 12(13), 1728. Bathina, S., Srinivas, N. and Das, U.N. 2017. Streptozotocin produces oxidative stress, inflammation and decreases BDNF concentrations to induce apoptosis of RIN5F cells and type 2 diabetes mellitus in Wistar rats. Biochem. Biophys. Res. Commun. 486(2), 406–13. Burillo, J., Marqués, P., Jiménez, B., González-Blanco, C., Benito, M. and Guillén, C. 2021. Insulin resistance and diabetes mellitus in Alzheimer’s disease. Cells 10(5), 1236. Cardinal, J.W., Margison, G.P., Mynett, K.J., Yates, A.P., Cameron, D.P. and Elder, R.H. 2001. Increased susceptibility to streptozotocin-induced β-cell apoptosis and delayed autoimmune diabetes in alkylpurine-dna-n-glycosylase-deficient mice. Mol. Cell Biol. 21(16), 5605–5613. Chen, G.F., Xu, T.H., Yan, Y., Zhou, Y.R., Jiang, Y., Melcher, K. and Xu, H.E. 2017. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 38(9), 1205–1235. Chen, Y., Liang, Z., Blanchard, J., Dai, C.L., Sun, S., Lee, M.H., Grundke-Iqbal, I., Iqbal, K., Liu, F. and Gong, C.X. 2013. A non-transgenic mouse model (icv-STZ mouse) of Alzheimer’s disease: similarities to and differences from the transgenic model (3xTg-AD mouse). Mol. Neurobiol. 47(2), 711–725. De Sousa, R.A.L., Harmer, A.R., Freitas, D.A., Mendonça, V.A., Lacerda, A.C.R. and Leite, H.R. 2020. An update on potential links between type 2 diabetes mellitus and Alzheimer’s disease. Mol. Bio. Rep. 47(8), 6347–6356. Deng, Y., Li, B., Liu, Y., Iqbal, K., Grundke-Iqbal, I. and Gong, C.X. 2009. Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain: Implication for Alzheimer’s disease. Am. J. Pathol. 175(5), 2089–2098. Deture, M.A. and Dickson, D.W. 2019. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegeneration 14(1), 1–18. Drummond, E. and Wisniewski, T. 2017. Alzheimer’s disease: experimental models and reality. Acta Neuropathol. 133(2), 155–175. Eleazu, C.O., Eleazu, K.C., Chukwuma, S. and Essien, U.N. 2013. Review of the mechanism of cell death resulting from streptozotocin challenge in experimental animals, its practical use and potential risk to humans. J. Diabetes Metab. Disord. 12(1), 60. Evans, D.B., Rank, K.B., Bhattacharya, K., Thomsen, D.R., Gurney, M.E. and Sharma, S.K. 2000. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase ii inhibits tau’s ability to promote microtubule assembly. J. Biol. Chem. 275(32), 24977–24983. Goud, B.J., Dwarakanath, V. and Swamy, B.K.C. 2015. Streptozotocin -a diabetogenic agent in animal models. Int J Pharm Pharm Res. 3(31), 253–269. Grieb, P. 2016. Intracerebroventricular streptozotocin injections as a model of Alzheimer’s disease: in search of a relevant mechanism. Mol. Neurobiol. 53(3), 1741–1752. Gupta, S., Yadav, K., Mantri, S.S., Singhal, N.K., Ganesh, S. and Sandhir, R. 2018. Evidence for compromised insulin signaling and neuronal vulnerability in experimental model of sporadic Alzheimer’s disease. Mol. Neurobiol. 55(12), 8916–8935. Jolivalt, C.G., Hurford, R., Lee, C.A., Dumaop, W., Rockenstein, E. and Masliah, E. 2010. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp. Neurol. 223(2), 422–431. Jope, R.S. and Johnson, G.V.W. 2004. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29(2), 95–102. Kleinridders, A., Ferris, H.A., Cai, W. and Kahn, C.R. 2014. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 63(7), 2232–43. Kothari, V., Luo, Y., Tornabene, T., O’Neill, A.M., Greene, M.W., Geetha, T. and Babu, J.R. 2017. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis Dis. 1863(2), 499–508. Lee, H.J., Seo, H.I., Cha, H.Y., Yang, Y.J., Kwon, S.H. and Yang, S.J. 2018. Diabetes and Alzheimer’s disease: mechanisms and nutritional aspects. Clin. Nutr. Res. 7(4), 229. Lee, M.S., Kao, S.C., Lemere, C.A., Xia, W., Tseng, H.C., Zhou, Y., Neve, R., Ahlijanian, M.K. and Tsai, L.H. 2003. APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 163(1), 83–95. Lester-Coll, N., Rivera, E.J., Soscia, S.J., Doiron, K., Wands, J.R. and De La Monte, S.M. 2006. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J. Alzheimers Dis. 9(1), 13–33. Liu, F., Li, B., Tung, E.J., Grundke-Iqbal, I., Iqbal, K. and Gong, C.X. 2007. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 26(12), 3429–36. Ma, Q.L., Yang, F., Rosario, E.R., Ubeda, O.J., Beech, W., Gant, D.J., Ping, P.C., Hudspeth, B., Chen, C., Zhao, Y., Vinters, H.V., Frautschy, S.A. and Cole, G.M. 2009. β-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-jun n-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J. Neurosci. 29(28), 9078–89. Murphy, M.P. and Levine, H. 2010. Alzheimer’s disease and the amyloid-β peptide. J. Alzheimers Dis. 19(1), 311–23. Oliveira, W.H., Braga, C.F., Lós, D.B., Araújo, S.M.R., França, M.E.R., Duarte-Silva, E., Rodrigues, G.B., Rocha, S.W.S. and Peixoto, C.A. 2021. Metformin prevents p-tau and amyloid plaque deposition and memory impairment in diabetic mice. Exp. Brain Res. 239(9), 2821–39. Orr, M.E., Sullivan, A.C. and Frost, B. 2017. A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol. Sci. 38(7), 637-48. Park, S.A. 2011. A common pathogenic mechanism linking type-2 diabetes and Alzheimer’s disease: Evidence from animal models. J. Clin. Neurol. 7(1), 10-8. Picone, P., Di Carlo, M. and Nuzzo, D. 2020. Obesity and Alzheimer’s disease: molecular bases. Eur. J. Neurosci. 52(8), 3944–3950. Pieper, A.A., Verma, A., Zhang, J. and Snyder, S.H. 1999. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol. Sci. 20(4), 171–181. Radenković, M., Stojanović, M. and Prostran, M. 2016. Experimental diabetes induced by alloxan and streptozotocin: the current state of the art. J. Pharmacol. Toxicol. Methods 78, 13–31. Rajmohan, R. and Reddy, P.H. 2017. Amyloid beta and phosphorylated tau accumulations cause abnormalities at synapses of Alzheimer’s disease neurons. J. Alzheimers Dis. 57(4), 975–999. Ramanathan, A., Nelson, A.R., Sagare, A.P. and Zlokovic, B.V. 2015. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer’s disease: the role, regulation and restoration of LRP1. Front. Aging Neurosci. 15(7), 136. Ravelli, K.G., Rosário, B. dos A., Camarini, R., Hernandes, M.S. and Britto, L.R. 2017. Intracerebroventricular streptozotocin as a model of Alzheimer’s disease: neurochemical and behavioral characterization in mice. Neurotox. Res. 31(3), 327–33. Reddy, P.H. and Beal, M.F. 2008. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 14(2), 45-53. Salkovic-Petrisic, M., Knezovic, A., Hoyer, S. and Riederer, P. 2013. What have we learned from the streptozotocin-induced animal model of sporadic Alzheimer’s disease, about the therapeutic strategies in Alzheimer’s research. J. Neural Transm.. 120(1), 233–252. Serrano-Pozo, A., Frosch, M.P., Masliah, E. and Hyman, B.T. 2011. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 1(1), a006189. Sevenich, L. 2018. Brain-resident microglia and blood-borne macrophages orchestrate central nervous system inflammation in neurodegenerative disorders and brain cancer. Front. Immunol. 6(9), 697. Šoltésová, D. and Herichová, I. 2011. On the mechanisms of diabetogenic effects of alloxan and streptozotocin. Diabetol. Metab. Endokrinol. 14(3), 130–113. Talbot, K., Wang, H.Y., Kazi, H., Han, L.Y., Bakshi, K.P., Stucky, A., Fuino, R.L., Kawaguchi, K.R., Samoyedny, A.J., Wilson, R.S., Arvanitakis, Z., Schneider, J.A., Wolf, B.A., Bennett, D.A., Trojanowski, J.Q. and Arnold, S.E. 2012. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Invest. 122(4), 1316–1338. Tomás Díaz-Gerevini, G., Daín, A., Pasqualini, M.E., López, C.B., Eynard, A.R. and Repossi, G. 2019. Diabetic encephalopathy: beneficial effects of supplementation with fatty acids ω3 and nordihydroguaiaretic acid in a spontaneous diabetes rat model. Lipids Health Dis. 18(1), 1–15. Ventura-Sobrevilla, J., Boone-Villa, V.D., Aguilar, C.N., Román-Ramos, R., Vega-Ávila, E., Campos-Sepúlveda, E. and Alarcón-Aguilar, F. 2011. Effect of varying dose and administration of streptozotocin on blood sugar in male CD1 mice. Proc. West Pharmacol. Soc. 54, 5–9. Wang, K., Song, F., Xu, K., Liu, Z., Han, S., Li, F. and Sun, Y. 2019. Irisin attenuates neuroinflammation and prevents the memory and cognitive deterioration in streptozotocin-induced diabetic mice. Mediators Inflamm. 2019, 1567179. Yang, Y., Ma, D., Wang, Y., Jiang, T., Hu, S., Zhang, M., Yu, X. and Gongb, C.X. 2013a. Intranasal insulin ameliorates tau hyperphosphorylation in a rat model of type 2 diabetes. J. Alzheimers Dis. 33(2), 329–338. Yang, Y., Wu, Y., Zhang, S. and Song, W. 2013b. High glucose promotes ab production by inhibiting app degradation. PLoS One 8(7), 69824. Yiannopoulou, K.G. and Papageorgiou, S.G. 2020. Current and future treatments in Alzheimer disease: an update. J. Cent. Nerv. Syst. Dis. 12, 117957352090739. Zhang, Y., Ding, R., Wang, S., Ren, Z., Xu, L., Zhang, X., Zhao, J., Ding, Y., Wu, Y. and Gong, Y. 2018. Effect of intraperitoneal or intracerebroventricular injection of streptozotocin on learning and memory in mice. Exp. Ther. Med. 16(3), 2375–2380. | ||
| How to Cite this Article |
| Pubmed Style Titisari N, Ahmad H, Samsulrizal N, Fauzi A, Razak ISA. The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model. Open Vet. J.. 2025; 15(2): 594-600. doi:10.5455/OVJ.2025.v15.i2.8 Web Style Titisari N, Ahmad H, Samsulrizal N, Fauzi A, Razak ISA. The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model. https://www.openveterinaryjournal.com/?mno=231126 [Access: June 26, 2026]. doi:10.5455/OVJ.2025.v15.i2.8 AMA (American Medical Association) Style Titisari N, Ahmad H, Samsulrizal N, Fauzi A, Razak ISA. The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model. Open Vet. J.. 2025; 15(2): 594-600. doi:10.5455/OVJ.2025.v15.i2.8 Vancouver/ICMJE Style Titisari N, Ahmad H, Samsulrizal N, Fauzi A, Razak ISA. The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model. Open Vet. J.. (2025), [cited June 26, 2026]; 15(2): 594-600. doi:10.5455/OVJ.2025.v15.i2.8 Harvard Style Titisari, N., Ahmad, . H., Samsulrizal, . N., Fauzi, . A. & Razak, . I. S. A. (2025) The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model. Open Vet. J., 15 (2), 594-600. doi:10.5455/OVJ.2025.v15.i2.8 Turabian Style Titisari, Nurina, Hafandi Ahmad, Nurdiana Samsulrizal, Ahmad Fauzi, and Intan Shameha Abdul Razak. 2025. The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model. Open Veterinary Journal, 15 (2), 594-600. doi:10.5455/OVJ.2025.v15.i2.8 Chicago Style Titisari, Nurina, Hafandi Ahmad, Nurdiana Samsulrizal, Ahmad Fauzi, and Intan Shameha Abdul Razak. "The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model." Open Veterinary Journal 15 (2025), 594-600. doi:10.5455/OVJ.2025.v15.i2.8 MLA (The Modern Language Association) Style Titisari, Nurina, Hafandi Ahmad, Nurdiana Samsulrizal, Ahmad Fauzi, and Intan Shameha Abdul Razak. "The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model." Open Veterinary Journal 15.2 (2025), 594-600. Print. doi:10.5455/OVJ.2025.v15.i2.8 APA (American Psychological Association) Style Titisari, N., Ahmad, . H., Samsulrizal, . N., Fauzi, . A. & Razak, . I. S. A. (2025) The mechanism underlying streptozotocin injection for the development of a nontransgenic Alzheimer’s disease animal model. Open Veterinary Journal, 15 (2), 594-600. doi:10.5455/OVJ.2025.v15.i2.8 |

