




| Research Article | ||
Open Vet. J.. 2025; 15(10): 5273-5283 Open Veterinary Journal, (2025), Vol. 15(10): 5273-5283 Research Article Molecular characterization of Hepatozoon canis in dogs from Can Tho in VietnamTran Thi Thao*, Nguyen Thanh Thien, Luu Dac Gia, Kha Thanh Thu, Dang Thi Tham, Nguyen Tran Phuoc Chien and Tran Ngoc BichFaculty of Veterinary Medicine, College of Agriculture, Can Tho University, Can Tho, Vietnam *Corresponding Author: Tran Thi Thao. Faculty of Veterinary Medicine, College of Agriculture, Can Tho University Campus II, 3/2 Street, Ninh Kieu district, Can Tho City, Vietnam. Email: ttthaoty [at] ctu.edu.vn Submitted: 24/02/2025 Revised: 22/08/2025 Accepted: 12/09/2025 Published: 31/10/2025 © 2025 Open Veterinary Journal
AbstractBackground: Hepatozoon canis (H. canis) is an intracellular protozoan parasite transmitted by ingestion of infected ticks. It is responsible for canine hepatozoonosis, a disease that affects the health and well-being of dogs worldwide. The infection can range from subclinical to severe, depending on the host’s immune status and parasite burden. Hepatozoon canis has been reported in multiple regions, but data on its prevalence and genetic characteristics in Vietnam, particularly in Can Tho City, remains limited. Aim: To determine the infection rate and genetic characteristics of H. canis in dogs in Can Tho City, Vietnam. Methods: A total of 359 suspected cases of H. canis presented to veterinary hospitals between January and November 2024 were investigated. Molecular diagnosis was performed using polymerase chain reaction, and positive samples were sequenced. The resulting sequences were compared with reference H. canis strains in GenBank to assess genetic similarity and variation. Results: The overall infection rate of H. canis was 0.75% and 9.47% among the surveyed dogs and suspected cases, respectively. Genetic analysis showed high similarity (98.88%–100%) between the three local and global reference strains. Four high-entropy loci were observed: 208 (G→A), 217 (T→C), 450 (C→T), and 474 (C→T), with the highest entropy at position 450 (0.682), suggesting regional genetic variation and potential selective pressure. Transition bias analysis revealed a strong preference for pyrimidine transitions (T↔C), indicating conservation in key regions. Conclusion: To the best of our knowledge, this is the first comprehensive genetic characterization of H. canis in dogs in Can Tho City. Despite a low prevalence of infection, genetic analysis revealed significant local variation. These findings enhance our understanding of the molecular epidemiology and evolution of H. canis and emphasize the need for further studies to elucidate the clinical implications of genetic polymorphisms. Keywords: Molecular epidemiology, Hepatozoon canis, Tick-borne pathogen, 18S rRNA gene, Vietnam. IntroductionHepatozoon canis, first identified in India in 1905, is a tick-borne apicomplexan protozoan that infects domestic and wild canids across diverse geographic regions, including Asia, Africa, and Europe (Baneth, 2011; Miterpáková et al., 2017). All Hepatozoon spp. share a basic lifecycle, involving sporogonic development in a hematophagous invertebrate host, followed by gametogenesis and sporulation in a vertebrate intermediate host (Baneth et al., 2007). Hepatozoon canis sporozoites spread via the blood and lymphatic system to several organs, including the spleen, bone marrow, lungs, liver, and kidneys, infecting leukocytes and parenchymal tissue cells (Vásquez-Aguilar et al., 2021). Hepatozoonosis generally manifests in a chronic form, with no established correlation between the age of affected dogs and H. canis infection. The geographic distribution of H. canis infection is closely associated with Rhipicephalus sanguineus (R. sanguineus), the most widely distributed tick species globally (Giannelli et al., 2013). In Vietnam, the prevalence of tick infestation in dogs was reported at 29.01%, with the brown dog tick (R. sanguineus) being the predominant species (Do et al., 2024), posing a risk for the spread of canine vector-borne diseases, especially under tropical conditions favorable for tick proliferation. Although H. canis has been reported in various countries, molecular and epidemiological data remain limited in Vietnam. According to Nguyen et al. (2021), the prevalence of H. canis in Ho Chi Minh City was 11.33%; however, comprehensive studies in the Mekong Delta have not yet been conducted. This study aimed to investigate the prevalence of H. canis among clinically suspected dogs and to genetically characterize local strains using partial 18S rRNA gene sequencing, given the lack of molecular data from Southern Vietnam, particularly in Can Tho City—a central hub of the Mekong Delta. These findings will help elucidate the genetic diversity and potential epidemiological patterns of H. canis in this region. Materials and MethodsStudy area and samplingDogs exhibiting clinical signs consistent with blood-borne parasitic infections—such as fever, anorexia, lethargy, emaciation, muscle atrophy, anemia, and pale mucous membranes—were initially examined at participating veterinary hospitals in Can Tho City from January to November 2024. The inclusion criteria encompassed dogs presenting with at least two of the aforementioned clinical signs during clinical examination. A total of 359 dogs that met these criteria were enrolled for further testing. Blood smear was examined on all enrolled dogs as an initial screening method to evaluate hemoparasites, although molecular testing was prioritized due to its limited sensitivity. Subclinical cases—dogs lacking observable symptoms—were excluded from this study. Whole blood (2 ml) was collected from each dog via cephalic venipuncture, placed in EDTA K3 anticoagulant tubes, stored in a refrigerated container, and transported to the Faculty of Veterinary Medicine, Can Tho University, for molecular analysis. Hematological data, such as red blood cell count, hemoglobin concentration, and platelet count, were not available for all animals; therefore, anemia was assessed based on mucous membrane pallor. Platelet counts were not systematically evaluated due to resource constraints. This clarification provides transparency regarding the inclusion criteria, screening methods, and limitations of hematological diagnostics. Molecular detection and characterization of H. canisGenomic DNA was isolated from whole blood samples of dogs using a TopPURE® RNA/DNA Viral Extraction Kit (ABT, Vietnam). The DNA sample was eluted in 50 μl of EB buffer, and the concentration of the pure DNA was determined using a NanoDrop™ 2000 spectrophotometer (Thermo Scientific™) by measuring the 260/280 and 260/230 ratios. The DNA was aliquoted at a concentration of 100 ng/μl. The aliquots were ultimately kept at −20°C for future use. The DNA samples were subjected to polymerase chain reaction (PCR) in a 25 µl reaction volume, including 12 µl of 2X Mix Tracking Dye (Phusa genomics, Vietnam), 1 µl each of forward and reverse primers at a concentration of 10 µM, 9 µl of PCR-grade water, and 2 µl of DNA template. The primers used for the PCR reaction were HepF (5′-ATACATGAGCAAAATCTCAAC-3′) and HepR (5′-CTTATTATTCCATGCTGCAG-3′), as described by Inokuma et al. (2002), yielding an anticipated product size of 666 bp. The thermal cycling parameters were as follows: initial denaturation at 95°C for 5 minutes, followed by 35 cycles of 95°C for 30 seconds, 57°C for 30 seconds, and 72°C for 90 seconds, and a final extension at 72°C for 5 minutes. The PCR amplicons were stained with 6X GelRed Loading Buffer with Tricolor (ABT, Vietnam) and visualized using 1.5% agarose gel electrophoresis under LED illumination, followed by photography. Nuclease-free water served as a negative control, and DNA Ladder (Phusa genomics, Vietnam) was used as a reference for determining the molecular mass of the PCR results. Phylogenetic analysisThe 18S rRNA amplicons (666 bp in length) were purified from the PCR products using the TopPURE® PCR/Gel DNA purification kit (ABT, Vietnam). The presence of 18S rRNA inserts was confirmed by Sanger sequencing at Can Tho University, Vietnam. All sequences were analyzed using BLAST (NCBI, http://www.ncbi.nlm.nih.gov/ BLAST) and deposited in GenBank. The 18S rRNA gene sequences were aligned using the MUSCLE algorithm, and the genetic inference was analyzed using a phylogenetic tree with some gap-free positions. Phylogenetic trees were reconstructed using the maximum likelihood (ML) method implemented in MEGA.X software. Bootstrap analysis with 1,000 repetitions was used to assess the branching pattern reliability of the ML trees. The evolutionary distances were analyzed using the Kimura-2 parameter model. The trees were created and labeled using the Interactive Tree of Life (https://itol.embl.de/). The nucleotide similarity was assessed using the STD v.1.3 software. The similarity of nucleotide sequences was evaluated using a sequence identity matrix in Bioedit v.7.0.5.3. Entropy analysis was used to ascertain the variability of the nucleotide sequences. The sequences were aligned and analyzed by the entropy [H (x)] plot using Bioedit software v.7.0.5.3. Statistical analysisDescriptive data analysis and chart generation were performed using Microsoft Excel 2019. The Chi-squared test was used to analyze the effect of gender (male and female), age (<6 months, 6 months–2 years, 2–5 years, and ≥5 years), breed (imported or indigenous), rearing method (free-range or confined), and coat phenotype (short or long-thick) on the positivity of dogs for Hepatozoon DNA. A p-value of p < 0.05 was considered statistically significant. Statistical analysis was performed using Minitab 21.0 software. Risk factors were assessed using odds ratio (OR) analysis with WinEpi software (http://www.winepi.net/uk/index.htm). Ethical approvalThis study was conducted on animals with natural infections. This study used diagnostic samples, and no animal experiments were conducted. Informed consent was obtained from the owners for their animals’ participation in this study. ResultsSurvey results on H. canis infection in dogsThe results in Table 1 show 34 cases of H. canis presence, accounting for 9.47% of suspected cases (34/359) and 0.75% of the surveyed population (34/4528). The infection rate of H. canis in dogs across the two surveyed locations was not statistically significant (p > 0.05), suggesting a uniform distribution of H. canis across Can Tho City, without significant geographic clustering. Table 1 shows that the prevalence of H. canis infection in the indigenous dog group was 12.99%, which was significantly higher than that in the foreign dog group (6.04%), with a statistically significant difference (p < 0.05). Additionally, the risk of H. canis infection in the indigenous dog group was 2.32 times higher than that in the foreign dog group (95% CI: 1.10–4.92). This might reflect increased environmental exposure among local breeds, consistent with their husbandry practices. Table 1. Prevalence of H. canis infection in relation to risk factors.
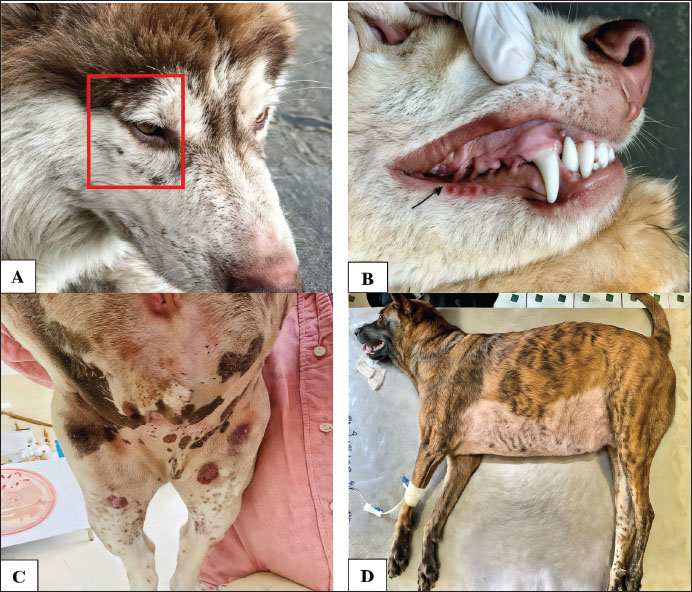
Furthermore, the study found that dogs with long and thick coat phenotypes had a prevalence of H. canis infection of 17.54%, higher than dogs with short coats (7.95%), with an increased OR of 2.47 (95% CI: 1.11–5.48; p < 0.05). The results in Table 1 also indicate that dogs aged ≥5 years had a prevalence of H. canis infection of 11.11%, although age was not a significant factor. This suggests that dogs of all ages can be infected with H. canis, a finding similar to observations regarding rearing methods and sex (p > 0.05). Among the 34 confirmed cases, the clinical presentations ranged from asymptomatic to severe. Frequently observed symptoms included lethargy, weakness, prolonged anemia, and cachexia. Additional findings, such as ataxia, elevated liver enzyme levels, generalized lymphadenopathy, and crusted skin lesions, were also recorded (Table 2). Figure 1 shows the representative manifestations, including tick infestation, subcutaneous hemorrhage, mucosal pallor, and respiratory distress.
Fig. 1. Clinical symptoms of H. canis infection in dogs. (A) Tick infestation in dogs infected with H. canis; (B) subcutaneous hemorrhage and pale mucous membranes in dogs infected with H. canis; (C) skin lesions in the abdominal region that are hemorrhagic; (D) fever and respiratory distress in dogs. Table 2. First report of H. canis infection in dogs and wolves in Can Tho: Clinical and epidemiological characteristics.
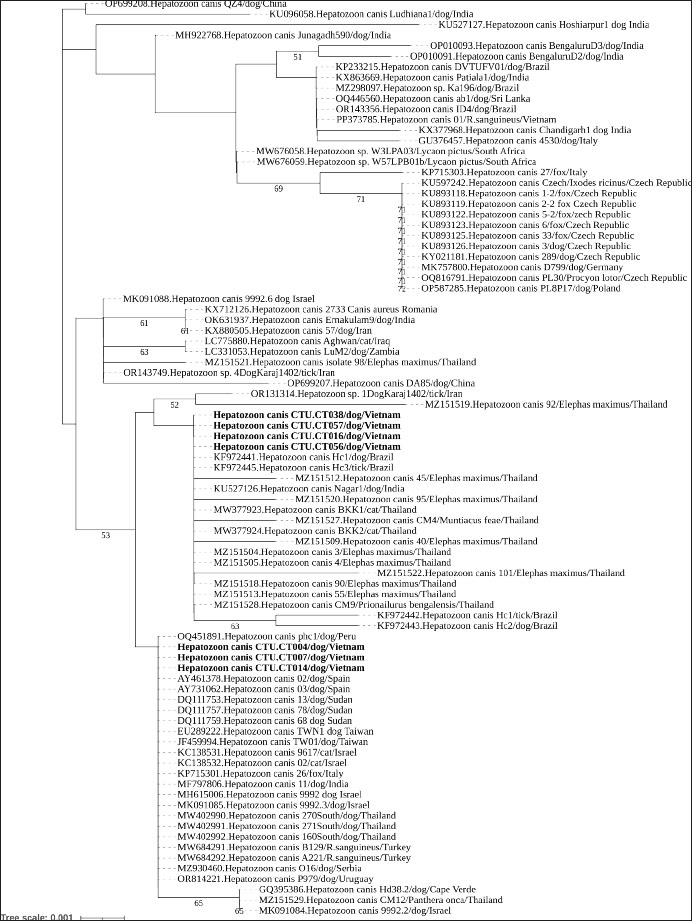
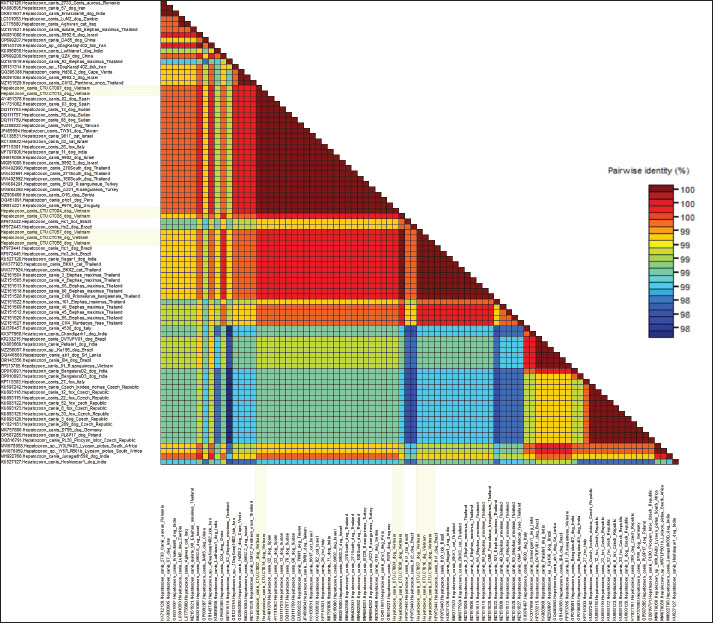
Genetic variation and phylogenetic analysis of 18S rRNAThis study further characterized H. canis by analyzing nearly complete 18S rRNA gene sequences from all PCR-positive dogs, which were subsequently deposited in GenBank under accession numbers PQ836033–PQ836039 (Table 2). The phylogenetic analysis of the 18S rRNA gene tree of H. canis was performed using MEGA.X. Generally, all H. canis sequences were divided into three well-supported clades, with >0.7% genetic divergence. The seven sequences identified in this study were clustered into two subclades of the first clade, which included global sequences (Fig. 2). Consistent with the genetic variation analysis, all of them shared the closest relationship with an identity of 98.3%–100% (Fig. 3). All seven sequences were closely related, sharing 99.8%–100% nucleotide identity and exhibiting high similarity with other known H. canis 18S rRNA gene variants (98.3%–100% identity) identified in various mammals and ticks.
Fig. 2. Phylogenetic position of H. canis isolates from dogs in Mekong Delta, Vietnam, Can Tho city based on 18S rRNA gene sequences The tree was constructed using the neighbor-joining Kimura-2 parameter method, and the tree numbers indicate bootstrap values for the branch points. The sequences identified from dogs in this study are marked in bold.
Fig. 3. Color-code matrix of pairwise identity scores generated by the alignment of nucleotide sequences of the gene 18S rRNA region of H. canis strains isolated in Can Tho city in Mekong Delta, Vietnam and representative isolates from other countries. Sequences identified from dogs in this study are marked in lemon yellow. Specifically, the sequences of strains CTU.CT004, CTU.CT007, and CTU.CT014 showed 100% nucleotide identity with partial sequences from dogs in Peru (OQ451891), Spain (AY461378, AY731062), Sudan (DQ111753, DQ111757, DQ111759), Israel (MH615006, MK091085), Uruguay (OR814221), Serbia (MZ930460), India (MF797806), Taiwan (EU289222, JF459994), Thailand (MW402990-MW402992), as well as sequences from H. canis isolated from cats in Israel (KC138531, KC138532), foxes in Italy (KP715301), and R. sanguineus ticks in Turkey (MW684291, MW684292). In contrast, the CTU.CT38, CTU.CT7, CTU.CT16, and CTU.CT56 strains demonstrated 99.6%–100% nucleotide similarity with the sequences isolated from dogs in Brazil (KF972441, KF972443) and India (KU527126). They also shared identity with sequences from isolates found in other animals, including cats (MW377923, MW377924), Prionailurus bengalensis (MZ151528), Muntiacus feae (MZ151527), and Elephas maximus (MZ151504, MZ151505, MZ151509, MZ151512, MZ151513, MZ151518, MZ151520, MZ151522) in Thailand, as well as ticks (KF972445, KF972442) in Brazil. These findings indicate the presence of at least two H. canis sublineages circulating in Can Tho. Furthermore, the studied strains were clustered into separate clades and shared 99.1%–99.3% nucleotide identity with the PP373785 strain isolated from R. sanguineus in Northern Vietnam, indicating intra-national genetic variability. However, the lowest nucleotide identity (98.3%–98.9%) was observed between sequences in this study and isolates from dogs in China (OP699208), India (KU096058, MH922768, and KU527127), and isolates from foxes (KY021181, KU893118–KU893126) in the Czech Republic. This reflects possible regional evolutionary divergence and adaptation. Each entry shows the substitution probability (r) from one base (row) to another base (column) (Kumar et al., 2018). The sum of the r values was normalized to 100. Transition substitutions were predominant, especially between pyrimidines (T↔C), which were notably high. Nucleotide frequencies were 27.68% (A), 32.80% (T/U), 23.31% (C), and 16.21% (G) (Table 3). The transition/transversion rate ratios were k1=0.138 (purines) and k2=30.585 (pyrimidines). The overall transition/transversion bias was R=9.519. This indicates a strong preference for transitions, especially T↔C, contributing to the genetic stability of the 18S rRNA gene. Table 3. This section presents the Maximum Composite Likelihood Estimate (MCLE) of nucleotide substitution patterns for the 18S rRNA gene of H. canis.
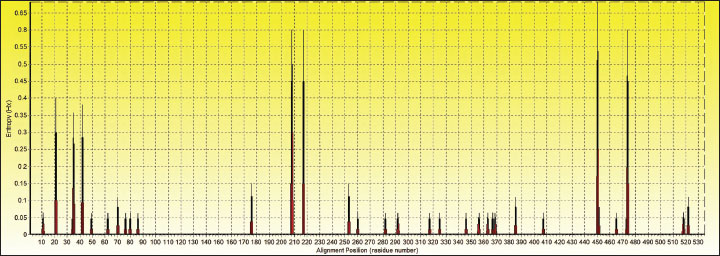
Entropy analysis of the nucleotide alignment of the H. canis 18S rRNA sequences (535 positions across 87 sequences) revealed 34 polymorphic sites with entropy values ranging from 0.063 to 0.682 (Fig. 4). Entropy values >0.4 indicate non-conserved positions. In this study, entropy values of 0.600–0.682 were observed at positions 208, 217, 450, and 474, with the highest value at position 450 (0.682). This high entropy value that while H. canis exhibits general genetic stability; certain sites may be under selective pressure or represent mutation hotspots.
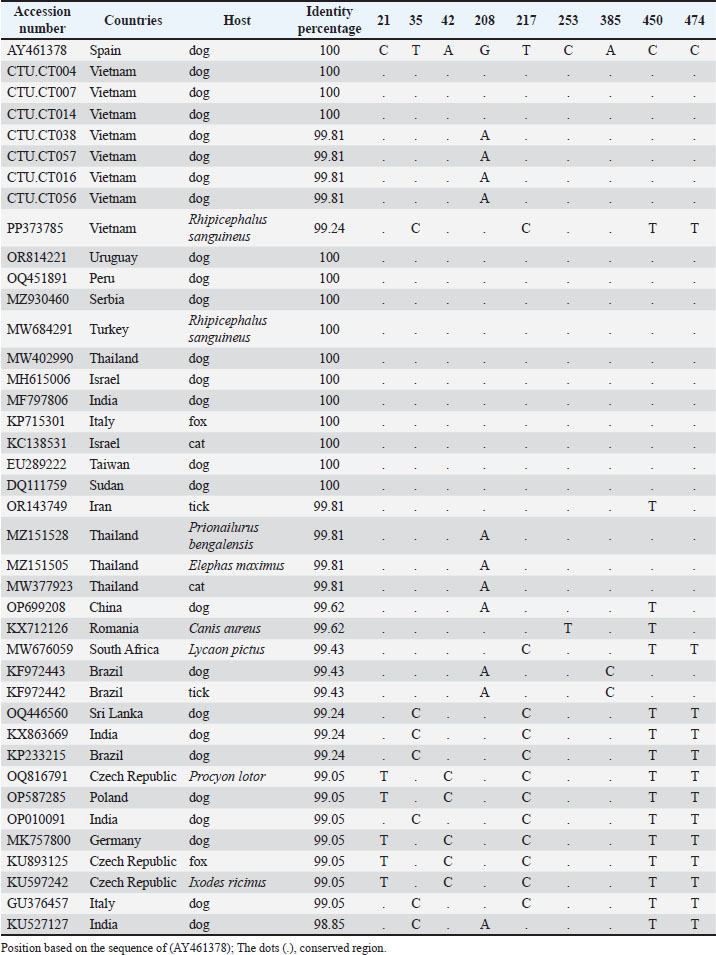
Fig. 4. Nucleotide sequences of partial 18S rRNA for H. canis isolates determined in this study were aligned with the published 18S rRNA sequences of the H. canis isolates in Spain (AY461378) as a reference and representative isolates from other countries. A dot indicates a conserved nucleotide relative to the published sequence. The nucleotide sequences of the partial 18S rRNA sequences of H. canis isolates from Can Tho (PQ836033–PQ836039) were aligned with a reference sequence from Spain (AY461378). The field strains CTU.CT38, CTU.CT7, CTU.CT16, and CTU.CT56 exhibited a nucleotide variation at position 208 (G→A), leading to a sequence similarity of 99.81% (534/535). This localized mutation may indicate genetic drift or regional adaptation among the Can Tho H. canis population. DiscussionThe findings of this study reinforce the widespread prevalence of H. canis in dogs in Vietnam and its association with several epidemiological factors. The absence of a significant relationship between infection status and age, sex, or rearing method aligns with the findings of previous studies (Licari et al., 2017; Çelik et al., 2022). These results indicate that H. canis infection is influenced by broader ecological factors, such as tick exposure, rather than confined to a particular demographic or management practice. Similarly, no association was found between rearing methods and infection rates, which may be attributed to the widespread presence of brown ticks in Vietnam (Do et al., 2024). Table 4. Alignment of partial sequences of 18S rRNA gene and position of variable sites.
One of the key risk factors identified in this study was breed, with indigenous dogs having a significantly higher likelihood of infection than imported ones. This finding supports previous reports that indigenous dogs are more exposed to tick-infested environments (Gomes et al., 2010; Revathi et al., 2022). The higher infection rates in Vietnam may be linked to the increased exposure to pathogens in indigenous breeds compared with exotic breeds, as indigenous dogs are often raised in more open environments, leading to higher exposure to ticks. Similarly, Do et al. (2024) reported that indigenous and mixed-breed dogs in Vietnam are more likely to be infested with ticks than exotic breeds. In addition, dogs with long and thick coats had a 2.47 times higher risk of contracting hepatozoonosis than dogs with short coats. Gomes et al. (2010) conducted a study in Brazil using a blood smear examination, which revealed a significant association between dog breeds and the prevalence of H. canis. Similarly, Revathi et al. (2022) surveyed 482 canine blood samples in the province of Chennai, Thailand. They reported higher prevalence rates of H. canis in long-haired and thick-coated breeds, such as Golden Retrievers (16.7%) and Cocker Spaniels (14.3%). Conversely, Gomes et al. (2010) observed lower infection rates in short-coated breeds, such as Pinschers and Pit Bulls (2.12%). This indicates that breed group and coat phenotype are risk factors for H. canis infection in Can Tho, Vietnam. The intensity of clinical symptoms resulting from H. canis infection correlated with the degree of parasitemia, leading to a spectrum of clinical manifestations ranging from subclinical in ostensibly healthy dogs to severe, potentially life-threatening conditions, with pronounced symptoms including extreme weakness, lethargy, and anemia (Mrljak et al., 2017; Kuručki et al., 2022). Clinical indications alone may lead to misdiagnoses because veterinary clinics seldom use H. canis DNA screening techniques. It is essential to develop rapid and highly sensitive pathogen detection methods using H. canis-specific antibodies or PCR. Diagnostic techniques are required to diagnose hepatozoonosis and immediately track its spread. The phylogenetic analysis revealed that our H. canis sequences clustered within the same group as other H. canis sequences obtained from dogs, foxes, jackals, cats, Prionailurus bengalensis, and Elephas maximus from Thailand, as well as various sequences from other regions worldwide. This clustering indicates the presence of a H. canis strain variant among the two different isolates. Furthermore, the analysis indicated that our most frequently observed H. canis sequence showed the highest similarity to H. canis sequences derived from golden jackals, supporting the previous hypothesis by Juwaid et al. (2019) that red foxes and jackals play a significant role in this pathogen’s distribution and transmission. Sequence comparisons of H. canis revealed that the isolates identified in both dogs and R. sanguineus shared 99.0%–100% similarity with the corresponding H. canis isolates from dogs in Spain and Croatia (Estrada-Peña et al., 2017; Uiterwijk et al., 2023). In particular, strains CTU.CT004, CTU.CT007, and CTU.CT014 from this study demonstrated 100% identity with isolates from Peru (OQ451891), Spain (AY461378), Sudan (DQ111753), and Israel (MH615006), suggesting the potential of genetically stable strains to be ancestrally conserved or to have recently been distributed globally. Conversely, isolates CTU.CT038, CTU.CT016, CTU.CT056, and CTU.CT057 showed 99.6%–99.8% identity with samples from Brazil (KF972443) and India (KU527126), indicating minor regional polymorphisms possibly driven by local vector or host dynamics. Previous studies have focused on the genetic diversity of H. canis in a particular country, indicating around 1% genetic variation (de Azevedo Gomes et al., 2016; Ahmad et al., 2018). In this study, all 18S rRNA gene sequences recovered from sick dogs exhibited the closest relationship to each other and other known sequences from large-scale mammals and ticks inside and outside Vietnam, as shown by nucleotide identity and phylogenetic analysis. This investigation revealed little genetic variation, as seen by the modest variance in the 18S rRNA sequences; nevertheless, the sequences from Vietnam, including those examined, had a nucleotide identity ranging from 98.3% to 100%, indicating some genetic diversity within H. canis. Although the overall nucleotide identity among global H. canis strains remains high (98.3%–100%), entropy analysis in this study revealed distinct polymorphic hotspots (position 450 with entropy 0.682), indicating that Vietnamese strains share core sequences with international counterparts and exhibit localized genetic signatures. This reflects the conservation of the 18S rRNA gene and the evolutionary divergence influenced by regional ecological pressures. Nucleotide changes in the 18S rRNA gene of H. canis across 87 global isolates revealed 34 polymorphic sites, with entropy values ranging from 0.063 to 0.682. High entropy positions were concentrated at loci 208 (G→A), 217 (T→C), 450 (C→T), and 474 (C→T), indicating regions under selective pressure. Notably, position 450 exhibited the highest entropy (0.682), indicating significant genetic variation. Nucleotide composition study showed that pyrimidines (T and C) outnumbered purines (A and G). The substitution patterns indicate a substantial predilection for transitions (R=9.519), especially between pyrimidines (T↔C), reflecting this composition. This transition/transversion bias affects evolutionary mechanisms that stabilize key 18S rRNA gene regions. The Vietnamese isolates were substantially conserved, correlating with Asian and African isolates, but the European and South American isolates had significant nucleotide alterations, indicating regional genetic divergence or adaptation to local host/vector dynamics. The presence of identical pathogen genotypes in different animal species indicates possible direct or horizontal transmission through these hosts (Hodžić et al., 2018; Helm et al., 2020). Finally, genomic data and molecular markers are critically required for the execution of successful methods to identify, regulate, and treatment of Hepatozoon infections. These findings are consistent with those of Vásquez-Aguilar et al. (2021), who emphasized the gene flow and population expansion of H. canis across continents. Our Vietnamese strains likely represent a part of this global genetic continuum, highlighting the importance of comparative molecular epidemiology to trace transmission routes and assess zoonotic potential. ConclusionOverall, the molecular procedures employed in this study successfully detected H. canis infections in domestic dogs in Can Tho City, Vietnam, and provided insights into their preliminary genetic diversity. This study provides a foundation for further research on the genetic and epidemiological aspects of H. canis in Vietnam and beyond. AcknowledgmentsThe authors would like to thank the technical staff at all veterinary medicine hospitals in Can Tho City for their invaluable assistance with sample collection. Special thanks to the Faculty of Veterinary Medicine, College of Agriculture, Can Tho University, for their support in sequencing and analyzing the samples. Additionally, the authors sincerely thank the dog owners in the region for their cooperation and willingness to participate in this study, which was instrumental in achieving their research objectives. Conflicts of interestThe authors have no conflicts of interest to declare. FundingThis research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors. Authors’ contributionsTran Thi Thao and Nguyen Tran Phuoc Chien equally contributed to this work. Data availabilityThe data presented in this study are available upon request from the corresponding author. ReferencesAhmad, A.S., Saeed, M.A., Rashid, I., Ashraf, K., Shehzad, W., Traub, R.J., Baneth, G. and Jabbar, A. 2018. Molecular characterization of Hepatozoon canis from farm dogs in Pakistan. Parasitol. Res. 117, 1131–1138; doi:10.1007/s00436-018-5790-1 Baneth, G. 2011. Perspectives on canine and feline hepatozoonosis. Vet. Parasitol. 181(1), 3–11; doi:10.1016/j.vetpar.2011.04.015 Baneth, G., Samish, M. and Shkap, V. 2007. Life cycle of Hepatozoon canis (Apicomplexa: adeleorina: Hepatozoidae) in the tick Rhipicephalus sanguineus and domestic dog (Canis familiaris). J. Parasitol. 93(2), 283–299; doi:10.1645/GE-494R.1 Çelik, B.A., Çelik, O.Y., Ayan, A., Yilmaz, A.B., Kilinç, O.O. and Ayan, O.O. 2022. A Molecular survey of Hepatozoon canis in dogs in the Siirt province of Turkey. Acta Vet. Brno. 91(3), 277–283; doi:10.2754/avb202291030277 De Azevedo Gomes, L., Moraes, P.H.G., Do Nascimento, L.D.C.S., O’Dwyer, L.H., Nunes, M.R.T., Rossi, A.D.R.P., Aguiar, D.C.F. and Goncalves, E.C. 2016. Molecular analysis reveals the diversity of Hepatozoon species naturally infecting domestic dogs in a northern region of Brazil. Ticks Tick. Borne. Dis. 7(6), 1061–1066; doi:10.1016/j.ttbdis.2016.09.008 Do, T., Bui, L.K., Umemiya-Shirafuji, R., Inpankaew, T., Hasan, T., Zafar, I., Ma, Z., Hang, L., Mohanta, U.K., Amer, M., El-Sayed, S.A.E.S., Xuan, X. and Kamyingkird, K. 2024. The detection of zoonotic microorganisms in Rhipicephalus sanguineus (brown dog ticks) from Vietnam and the frequency of tick infestations in owned dogs. Front. Vet. Sci. 11, 1435441; doi:10.3389/fvets.2024.1435441 Estrada-Peña, A., Roura, X., Sainz, A., Miró, G. and Solano-Gallego, L. 2017. Species of ticks and carried pathogens in owned dogs in Spain: results of a one-year national survey. Ticks Tick. Borne. Dis. 8(4), 443–452; doi:10.1016/j.ttbdis.2017.02.001 Giannelli, A., Ramos, R.A.N., Di Paola, G., Mencke, N., Dantas-Torres, F., Baneth, G. and Otranto, D. 2013. Transstadial transmission of Hepatozoon canis from larvae to nymphs of Rhipicephalus sanguineus. Vet. Parasitol. 196(1-2), 1–5; doi:10.1016/j.vetpar.2013.02.017 Gomes, P.V., Mundim, M.J.S., Mundim, A.V., De Ávila, D.F., Guimarães, E.C. and Cury, M.C. 2010. Occurrence of Hepatozoon sp. in dogs in the urban area originating from a municipality in southeastern Brazil. Vet. Parasitol. 174(1-2), 155–161; doi:10.1016/j.vetpar.2010.07.020 Helm, C.S., Von Samson-himmelstjerna, G., Liesner, J.M., Kohn, B., Müller, E., Schaper, R., Pachnicke, S., Schulze, C. and Krücken, J. 2020. Identical 18S rRNA haplotypes of Hepatozoon canis in dogs and foxes in Brandenburg, Germany. Ticks Tick. Borne. Dis. 11(6), 101520; doi:10.1016/j.ttbdis.2020.101520 Hodžić, A., Alić, A., Beck, R., Beck, A., Huber, D., Otranto, D., Baneth, G. and Duscher, G.G. 2018. Hepatozoon martis n. sp.(Adeleorina: hepatozoidae): morphological and pathological features of a Hepatozoon species infecting martens (family Mustelidae). Ticks Tick. Borne. Dis. 9(4), 912–920; doi: 10.1016/j.ttbdis.2018.03.023 Inokuma, H., Okuda, M., Ohno, K., Shimoda, K. and Onishi, T. 2002. Analysis of the 18S rRNA gene sequence of a Hepatozoon detected in two Japanese dogs. Vet. Parasitol. 106(3), 265–271; doi:10.1016/S0304-4017(02)00065-1 Juwaid, S., Sukara, R., Penezić, A., Mihaljica, D., Veinović, G., Kavallieratos, N.G., Ćirović, D. and Tomanović, S. 2019. First evidence of tick-borne protozoan pathogens, Babesia sp. and Hepatozoon canis, in red foxes (Vulpes vulpes) in Serbia. Acta Vet. Hung. 67(1), 70–80; doi:10.1556/004.2019.008 Kumar, S., Stecher, G., Li, M., Knyaz, C. and Tamura, K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35(6), 1547–1549; doi:10.1093/molbev/msy096 Kuručki, M., Tomanović, S., Sukara, R. and Ćirović, D. 2022. High prevalence and genetic variability of Hepatozoon canis in grey wolf (Canis lupus L. 1758) population in Serbia. Animals 12(23), 3335; doi:10.3390/ani12233335 Licari, E., Takács, N., Solymosi, N. and Farkas, R. 2017. First detection of tick-borne pathogens of dogs from Malta. Ticks Tick. Borne. Dis. 8(3), 396–399; doi:10.1016/j.ttbdis.2017.01.002 Miterpáková, M., Antolová, D., Ondriska, F. and Gál, V. 2017. Human Dirofilaria repens infections diagnosed in Slovakia in the last 10 years (2007–2017). Wien. Klin. Wochenschr. 129, 634–641; doi:10.1007/s00508-017-1233-8 Mrljak, V., Kuleš, J., Mihaljevi., Torti, M., Gotić, J., Crnogaj, M., Živičnjak, T., Mayer, I., Šmit, I., Bhide, M. and Barić Rafaj, R. 2017. Prevalence and geographic distribution of vector-borne pathogens in apparently healthy dogs in Croatia. Vector. Borne. Zoonotic Dis. 17(6), 398–408; doi:10.1089/vbz.2016.1990 Nguyen, T.L.A., Ngo, D.D. and Du, T.V. 2021. Survey on blood parasite infections in domesticated dogs in Ho Chi Minh City. J. Agric. Rural Dev. (Vietnam) 02(08), 104–110; Available via https://scholar.dlu.edu.vn/thuvienso/bitstream/DLU123456789/155280/1/CVv201S162021104.pdf Revathi, P., Bharathi, M.V., Madhanmohan, M., Latha, B.R. and Rani, K.V. 2022. Molecular epidemiology, characterisation of Hepatozoon canis in dogs as well as in ticks and haemato-biochemical profile of the infected dogs in Chennai. Indian J. Anim. Res. 1, 1–8; doi:10.18805/IJAR.B-4801 Uiterwijk, M., Vojta, L., Šprem, N., Beck, A., Jurković, D., Kik, M., Duscher, G.G., Hodžić, A., Reljić, S., Sprong, H. and Beck, R. 2023. Diversity of Hepatozoon species in wild mammals and ticks in Europe. Parasites Vectors 16(1), 27; doi:10.1186/s13071-022-05626-8 Vásquez-Aguilar, A.A., Barbachano-Guerrero, A., Angulo, D.F. and Jarquín-Díaz, V.H. 2021. Phylogeography and population differentiation in Hepatozoon canis (Apicomplexa: hepatozoidae) reveal expansion and gene flow in world populations. Parasites Vectors 14(1), 1–14; doi:10.1186/s13071-021-04924-x | ||


| How to Cite this Article |
| Pubmed Style Thao TT, Thien NT, Gia LD, Thu KT, Tham DT, Chien NTP, Bich TN. Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam. Open Vet. J.. 2025; 15(10): 5273-5283. doi:10.5455/OVJ.2025.v15.i10.44 Web Style Thao TT, Thien NT, Gia LD, Thu KT, Tham DT, Chien NTP, Bich TN. Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam. https://www.openveterinaryjournal.com/?mno=244413 [Access: January 26, 2026]. doi:10.5455/OVJ.2025.v15.i10.44 AMA (American Medical Association) Style Thao TT, Thien NT, Gia LD, Thu KT, Tham DT, Chien NTP, Bich TN. Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam. Open Vet. J.. 2025; 15(10): 5273-5283. doi:10.5455/OVJ.2025.v15.i10.44 Vancouver/ICMJE Style Thao TT, Thien NT, Gia LD, Thu KT, Tham DT, Chien NTP, Bich TN. Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam. Open Vet. J.. (2025), [cited January 26, 2026]; 15(10): 5273-5283. doi:10.5455/OVJ.2025.v15.i10.44 Harvard Style Thao, T. T., Thien, . N. T., Gia, . L. D., Thu, . K. T., Tham, . D. T., Chien, . N. T. P. & Bich, . T. N. (2025) Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam. Open Vet. J., 15 (10), 5273-5283. doi:10.5455/OVJ.2025.v15.i10.44 Turabian Style Thao, Tran Thi, Nguyen Thanh Thien, Luu Dac Gia, Kha Thanh Thu, Dang Thi Tham, Nguyen Tran Phuoc Chien, and Tran Ngoc Bich. 2025. Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam. Open Veterinary Journal, 15 (10), 5273-5283. doi:10.5455/OVJ.2025.v15.i10.44 Chicago Style Thao, Tran Thi, Nguyen Thanh Thien, Luu Dac Gia, Kha Thanh Thu, Dang Thi Tham, Nguyen Tran Phuoc Chien, and Tran Ngoc Bich. "Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam." Open Veterinary Journal 15 (2025), 5273-5283. doi:10.5455/OVJ.2025.v15.i10.44 MLA (The Modern Language Association) Style Thao, Tran Thi, Nguyen Thanh Thien, Luu Dac Gia, Kha Thanh Thu, Dang Thi Tham, Nguyen Tran Phuoc Chien, and Tran Ngoc Bich. "Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam." Open Veterinary Journal 15.10 (2025), 5273-5283. Print. doi:10.5455/OVJ.2025.v15.i10.44 APA (American Psychological Association) Style Thao, T. T., Thien, . N. T., Gia, . L. D., Thu, . K. T., Tham, . D. T., Chien, . N. T. P. & Bich, . T. N. (2025) Molecular characterization of Hepatozoon canis in dogs from Can Tho in Vietnam. Open Veterinary Journal, 15 (10), 5273-5283. doi:10.5455/OVJ.2025.v15.i10.44 |

