




| Research Article | ||
Open Vet. J.. 2025; 15(10): 5312-5325 Open Veterinary Journal, (2025), Vol. 15(10): 5312-5325 Research Article Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreakBejan A. Dizayee1 and Evan L. Khlef2*1Central Veterinary Laboratory in Kurdistan Region, Erbil, Iraq 2Department of Physiology and Microbiology, College of Medicine, Hawler Medical University, Erbil, Iraq *Corresponding Author: Evan L. Khlef. Department of Physiology and Microbiology, College of Medicine, Hawler Medical University, Erbil, Iraq. Email: evan.latef [at] hmu.edu.krd Submitted: 15/04/2025 Revised: 07/09/2025 Accepted: 29/09/2025 Published: 31/10/2025 © 2025 Open Veterinary Journal
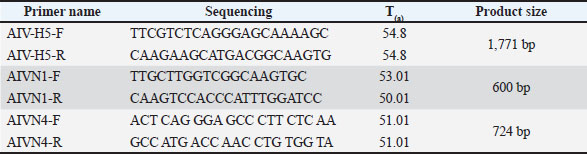
AbstractBackground: Influenza viruses continuously threaten avian and mammalian species. After the circulation of highly pathogenic avian influenza (HPAI) H5N1 in 2015 in Iraq, an H5N4 influenza virus emerged in domestic chickens. Aim: This study aimed to identify and characterize the presence of avian influenza H5N4 viruses in domestic poultry during the HPAI H5N1 circulation in 2015 in Iraq. Methods: Polymerase chain reaction, Sanger sequencing, and bioinformatics analysis were performed on the hemagglutinin and neuraminidase (NA) genotypes isolated from chickens in Iraq in 2015. Results: Out of 13 samples, eight (62%) were positive for the H5 gene, nine (69%) were positive for the NA gene, eight for N1, and one for N4. One sample, KCVL15-013, initially amplified using N1-specific primers, was later confirmed by sequencing to contain the N4 gene, likely due to partial primer homology. Sequence annotation confirmed the subtype as H5N4. Cleavage site analysis revealed a multibasic motif (RRRKR/GLF), indicating high pathogenicity. Phylogenetic analysis of the N4 gene showed that KCVL15-013 formed a distinct branch, clustering only with another Iraqi chicken-origin H5N4 strain, with weak bootstrap support (37%), and diverging from globally clustered wild bird-origin N4 viruses. Conclusion: This study concludes that H5N4 was co-detected during an H5N1 outbreak, indicating the concurrent circulation of multiple influenza A virus subtypes in poultry. Multiple strain genotypes of the influenza virus A could cause a co-infection in domestic chickens, particularly during a pandemic. Incorporating DNA sequencing and annotation into routine surveillance is essential for accurate detection of strain genotype. Keywords: Avian influenza, H5N4, HPAI, Sanger sequencing, NCBI. IntroductionInfluenza viruses are related to the Orthomyxoviridae family. This family includes negative-sense, single-stranded RNA viruses with a segmented genome (Arbeitskreis Blut, 2009). Influenza viruses are categorized into groups A, B, C, and D based on variation in their matrix proteins, internal nucleoproteins, and antigenic characteristics (Dadonaite et al., 2019). Influenza viruses are classified into subtypes based on the antigenic properties of the surface glycoproteins hemagglutinin (HA, H1-H18) and neuraminidase (NA, N1-N11), some of which show a significant threat to public health (Senne et al., 1996). Among the four influenza virus types (A, B, C, and D), influenza type A is a zoonotic disease with a complete natural reservoir in avian. Avian influenza virus can infect dogs, swine, horses, and humans, a disease caused by influenza virus strains that can be adapted to different species (Ghedin et al., 2005). Avian influenza viruses AIV are divided into highly pathogenic avian influenza (HPAI) and low pathogenic avian influenza (LPAI) based on the severity of the infection in birds (Neumann et al., 2007). HPAI is the more dangerous type, especially in poultry, where it can cause sudden illness and death, sometimes killing entire flocks. These viruses commonly are the H5 and H7 subtypes. While LPAI usually causes mild symptoms, or none at all, it is rarely fatal. Some LPAI viruses, particularly H5 and H7 strains, can change over time and become highly pathogenic. One of the key differences between HPAI and LPAI lies in the structure of the virus’s HA protein. HPAI viruses have a feature called a polybasic cleavage site, which enables them to spread throughout the bird’s entire body. In contrast, LPAI viruses have a simpler form that keeps the infection limited to the bird’s respiratory and digestive systems. When it comes to humans, some HPAI strains like H5N1 and H5N8 have infected people and caused concern for public health. LPAI viruses do not usually infect humans. From an economic standpoint, HPAI outbreaks can be devastating for the poultry industry due to the high death rates and strict control measures required. That is why monitoring both types of viruses is essential for protecting both animal and human health (WOAH/OIE, 2023; Alexander, 2007; Suarez, 2000). Numerous specific subtypes, 16 HA subtypes from H1 to H16 and nine NA subtypes from N1 to N9, should belong to the avian flu viruses, but the H17N10 and H18N11 subtypes, which are found in bats only (Capua and Alexander, 2004). The broad distribution of avian influenza viruses in different hosts may result in the exchange of their genes or gene fragments, contributing to highly antigenic variation and the potential development of new strains of avian influenza viruses (Dugan et al., 2008; Venkatesh et al., 2018a,b). This can cause significant epidemics and outbreaks, lead to economic losses in the poultry industry, and pose a threat to human health (Senne et al., 1996). The HA gene plays a crucial role in the life cycle of influenza virus A, specifically in the recognition of receptors, attachment of virus particles to the host cell, fusion of the virus with the host cell membrane, and entry into the host cell (De Graaf and Fouchier, 2014). Influenza A H5N1 was initially isolated from a Chinese goose in 1996. Human infections were first recorded in Hong Kong in 1997 (Brown et al., 2009). Asian H5N1 viruses were first detected in domestic geese in southern China in 1996. By 2000, their host range had expanded to include domestic ducks, which played a crucial role in the genesis of the 2003/2004 outbreaks. The epidemic was not due to the introduction and spread of a single virus, but it was caused by multiple viruses that were genotypically linked to the Goose/GD/96 lineage via the haemagglutinin gene. The H5N1 viruses isolated from China, including those from Hong Kong, between 1999 and 2004, had a range of genotypes and considerable variability within genotypes. The rising incidence and widespread reporting of disease in 2003/2004 can probably be attributed to the increasing spread of the viruses from existing reservoirs of infection in domestic waterfowl and live bird markets, leading to more significant environmental contamination. In Iraq, the virus struck a commercial farm near the Baghdad area, killing 13,240 birds out of 29,000. Avian influenza viruses with high pathogenicity have been identified in several Iraqi governorates over the past few years, resulting in significant economic losses for the poultry industry and posing a potential risk to human health (OIE, 2018). In 2006, the first report of HPAI subtype H5N1 in Iraq was from humans in Sulaymaniyah Governorate, Kurdistan region (CIDRAP, 2006). An isolated H5N1 outbreak was detected in backyard poultry in Sulaymaniyah Governorate, as reported by the Ministry of Agriculture in Kurdistan Region in May 2015 (Rashid et al., 2017; Saeed et al., 2021). This is the first study to analyze the genetic characteristics of the Iraqi H5N4 viruses. H5N4 is a novel subtype of HPAI. The appearance of H5N4 as HPAI highlights its potential to adapt and spread, particularly in domestic poultry populations. While no confirmed human infections have been reported, the zoonotic potential of H5N4 cannot be excluded, particularly given its shared ancestry with subtypes like H5N1, which have caused human illness. The lack of comprehensive data on H5N4, both in terms of molecular characteristics and epidemiological impact, underscores the need for ongoing surveillance and genetic analysis to assess its threat to both animal and human health (King et al., 2022; Xie et al., 2025). Continuous surveillance of HPAI H5N4 in Iraq is necessary to avoid health threats and financial losses (Saeed et al., 2021). To the best of our knowledge, this is the first report describing the molecular characterization of HPAI H5N4 in Iraq. Materials and MethodsSamples collectionDuring the outbreak of the avian influenza virus in Kurdistan, Iraq (Erbil, Sulaymaniyah, and Duhok Governorates) from May to December 2015, 13 samples were collected from chickens. Organ tissues (lungs, liver, intestine, spleen, heart, kidney, and brain) and cloacal and tracheal swab samples were obtained from the above-mentioned. Sample handling and RNA isolationSamples were analyzed and diagnosed in the Central Veterinary Laboratory in the Kurdistan Region. Swabs and biopsies from the same bird were considered one sample. Tissue samples were homogenized and pooled together. They were directed to RNA extraction. The Qiagen RNA extraction kit (RNeasy Mini Kit, QIAGEN, Germany) was used. According to the kit instructions, the RNA extraction was performed. The RNA was eluted in 50 µl of ddH2O. The samples were stored at −80°C until complementary DNA (cDNA) synthesis was performed. cDNA synthesisThe cDNA was synthesized by an M-Mulv kit (New England Biolabs, UK). According to the kit instructions, cDNA was synthesized for one reaction with a total volume of 25 μl by mixing buffer, forward primer, MgCl2, RNA, dNTPs, and enzyme. The volume was adjusted to 25 μl by adding ddH2O. The polymerase chain reaction (PCR) was programmed for one cycle at 42°C for 1 hour. After completing the PCR for cDNA synthesis, the product was stored at −80°C. PCR amplificationInvitrogen Platinum Taq polymerase (ThermoFisher Scientific, USA) was used. The PCR cocktail for the H5, N1, and N4 genes was prepared. The reaction was prepared with a total volume of 50 μl by mixing buffer, forward and reverse primers, MgCL2, template (cDNA from H5, N1, and N4), dNTPs, enhancer buffer, and enzyme. The volume was adjusted to 50 μl by adding ddH2O. The PCR was programmed for 35 cycles, including a 94°C pre-denaturation step for 30 seconds, 94°C denaturation step for 10 seconds, and 74°C extension step for 4 minutes. The annealing temperatures were varied depending on the amplified gene. For H5 amplification, the annealing temperature was 54.8°C for 30 seconds; for N1, the annealing temperature was 50°C for 30 seconds; while for N4, it was 51.01°C for 30 seconds (Table 1). The cycles were finished with the final extension step at 74°C for 10 minutes. Table 1. Primers used in this study.
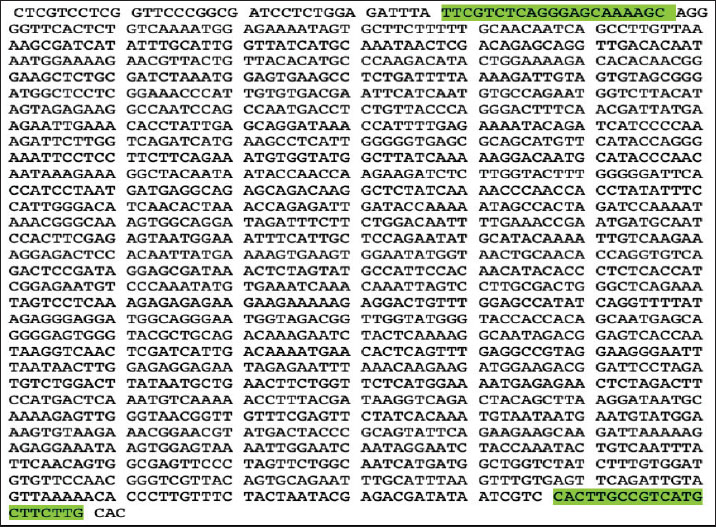
Agarose gel electrophoresisThe PCR products were gel electrophoresed using 1% agarose gel concentration. Agarose gel was prepared by dissolving 1 g of agarose powder in 100 ml of 1X TBE buffer (pH 8). Boiled and cooled it at room temperature (RT). A 1% agarose gel was prepared by incorporating 10 μl of ethidium bromide (10 mg/ml; Sigma-Aldrich, Germany) into 100 ml of molten agarose solution, resulting in a final concentration of 1 μg/ml. This concentration is slightly higher than the standard recommendation (~0.5 μg/ml) described by Sambrook (1989) and was used to enhance DNA band visibility during electrophoresis. The mixture was poured into the tray (8 × 8 cm) of the gel electrophoresis apparatus and left to solidify at RT. The samples were loaded into the agarose gel wells after mixing 6 μl of the sample with 2 μl of loading dye. The gel electrophoresis tank was run for 1 hour at 80, 10 V/cm. Then, the gel was placed on a UV transilluminator (Analytika Jena, USA), and DNA bands were detected by irradiation with UV light. The position of DNA bands on the gel is recorded using an image analyzer (a computerized digital camera) to check and analyze the results (Sambrook, 1989). Gel purificationThe positive DNA bands were extracted from the agarose gel using a QIAquick Gel Purification Kit, and the procedure followed the kit’s instructions. Sanger dideoxy sequencingThe gel-purified PCR products were sequenced using the Sanger dideoxy method by gene-specific primers at GENEWIZ Company, USA. The forward primers for H5, N1, and N4 were used for the sequencing process (Table 1). The gel-purified samples were dehydrated by a vacuum centrifuge (Eppendorf, Germany). The dried samples were well packed and transported to NJ, USA, with the DHL Express company. After a week, the results were received via email. Bioinformatics and data analysisSequencing data analysisThe sequencing data received from Genewiz were in the form of FASTA files with trimmed, ready-to-use data. The sequences were manually rechecked to be free from ambiguous bases (Ns) and the primer sequence. To confirm the viral subtype and examine the molecular characteristics, we utilized the NCBI Influenza Virus Annotation Tool, a specialized platform for identifying and analyzing influenza gene segments. Evolutionary Genetics Analysis (MEGA 12) software was used to construct phylogenetic trees and assess the evolutionary relationship between the Iraqi H5N4 strain and other global isolates. Primer designFor designing the H5 primers, the Influenza A virus [A/great crested grebe/Qinghai/1/2010 (H5N1)] with accession number HQ020376 is used as a control, as shown in Fig. 1. The NCBI N1 sequence with accession number EU930982.2 was used as a control for designing the N1 primers, as shown in Figure 2, while the NCBI N4 sequence with accession number CY096674.1 was used as a control for designing the N4 primers, as shown in Figure 3. The primer sequence and their annealing temperatures are provided in Table 1. The NCBI Primer-BLAST tool was used for designing the primers.
Fig. 1. Control H5 was used for primer design. The NCBI accession number is HQ020376.
Fig. 2. Control N1 was used for primer design. The NCBI accession number is EU930982.2.
Fig. 3. Control N4 was used for primer design. The NCBI accession number is CY096674. HA gene data analysisThe HA gene sequencing data for positive samples were analyzed using the NCBI annotation tool to confirm the HA gene and H5 genotype, as shown in Figure 8.
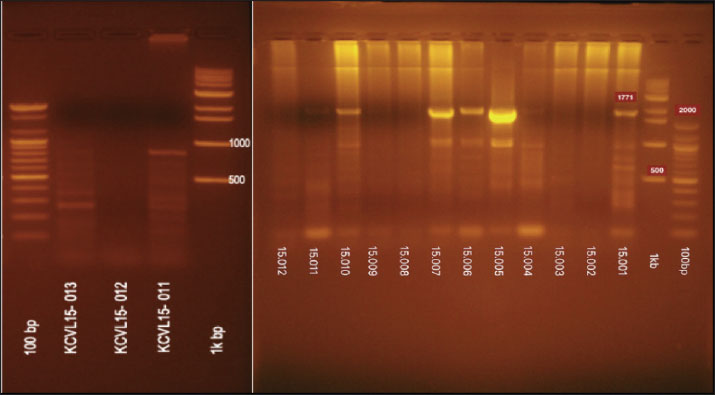
Fig. 4. The PCR results of the HA gene H5 subtype.
Fig. 5. The PCR result of re-amplification of KCVL15-013 of the HA gene H5 subtype.
Fig. 6. The PCR products for the N1 and the N4 genotypes using N1 primers. The last line of the PCR product (KCVL15-013) is the N4 genotype.
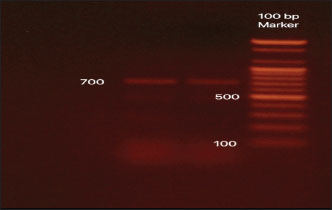
Fig. 7. The PCR amplification of the N4 subtype using specific primers.
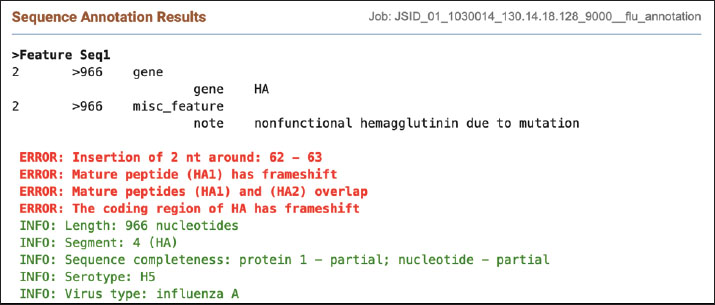
Fig. 8. Result of annotation of the H5 sequence. NA gene data analysisThe NCBI annotation tool was used to confirm the presence of the N1 and N4 genotypes. The results of the annotation are shown in Figures 9 and 10.
Fig. 9. Result of annotation of the N1 sequence.
Fig. 10. Result of annotation of the N4 sequence. HA pathogenesis identificationThe identification of the HPAI was based on the amino acid sequence. Specifically, after translating the HA nucleotides into an amino acid sequence, we sought to identify the cleavage site. The activation of the HA protein needs the host enzyme to cleave it into HA1 and HA2 subunits. The amino acid sequence at the HA cleavage site determines which host proteases can do this. This site is located right before the HA2 region begins. In low-pathogenic strains, the cleavage site usually contains just one basic amino acid, either arginine (R) or lysine (K). Because of this, the virus can only be activated by specific enzymes (trypsin-like proteases) that are found in limited areas of the body, like the respiratory or digestive tract. An example of this type of cleavage site is PQRETR↓GLF, which typically results in a mild or localized infection. However, in highly pathogenic strains, the cleavage site contains four or more basic amino acids in a motif. This "multibasic" site can be recognized by furin-like proteases, which are found ubiquitously in the body. As a result, the virus can spread much more easily and infect multiple organs, often leading to severe disease or death. A typical example of an HPAI cleavage site is PQRERRRKR↓GLF. By translating the HA gene into an amino acid sequence and checking the multibasic amino acid cleavage site, researchers can determine whether an avian influenza virus is likely to be a highly pathogenic strain (FAO, 2022). The HA gene sequencing data were translated to coding amino acids by the NCBI ORF finder. The amino acid of the HA gene cleavage site was detected, as shown in Fig. 11.
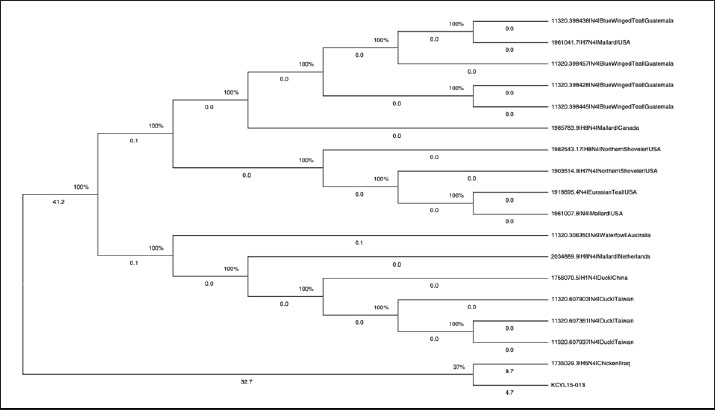
Fig. 11. The HA cleavage site showing the RRRKR/GLF motif indicating HPAI. Phylogenetic analysisTo determine the evolutionary relationship of the KCVL15-013 strain with other N4 subtype influenza viruses detected in 2015, a phylogenetic analysis was conducted using the MEGA (Molecular Evolutionary Genetics Analysis) software. All available nucleotide sequences of the H×N4 subtype of Influenza Virus A were collected in 2015. They were aligned with the KCVL15-013 sequence using the Clustal Omega algorithm, as implemented in MEGA. Then, a maximum likelihood phylogenetic tree was constructed based on the aligned sequences, employing the Tamura and Nei (1993) substitution model with 500 bootstrap replicates to assess the robustness of the tree topology. The resulting tree provided insight into the genetic relationship and evolutionary divergence of KCVL15-013 compared to other N4 subtype strains, as shown in Fig. 12 (Tamura and Nei, 1993; Al-Ouqaili et al., 2020; Sudhir Kumar et al., 2024).
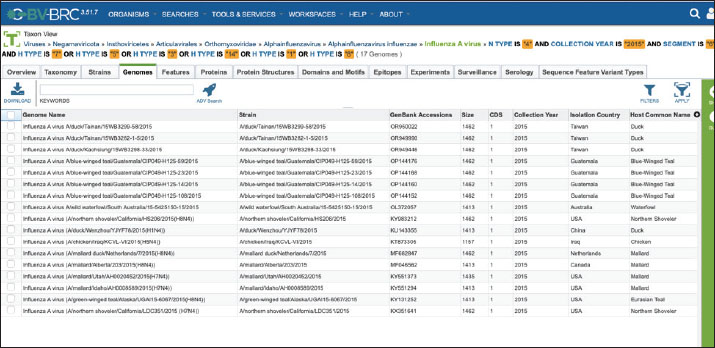
Fig. 12. Maximum likelihood phylogenetic tree (MEGA 12) of NA (N4) genes from influenza A viruses detected in 2015, including the KCVL15-013 strain. Bootstrap support values are displayed as percentages at internal nodes. Branch lengths reflect the number of nucleotide substitutions per site. Collecting the data of H×N4 subtype sequences of Influenza Virus A in 2015The H×N4 subtype sequences of Influenza A Viruses detected in 2015 were collected using the Bacterial and Viral Bioinformatics resource Center (BV-BRC) platform. The database was filtered specifically for the Influenza A virus, with the NA subtype set to N4 and the collection year limited to 2015. Additional filters were applied to the platform to extract metadata, including host species, geographic origin, and HA subtype. The selected sequences were then downloaded in FASTA format for alignment and phylogenetic analysis, as shown in Fig. 13.
Fig. 13. Dataset of HxN4 Influenza A virus sequences collected in 2015 using the BV-BRC platform. Ethical approvalThis study received approval from the Hawler Medical University/College of Medicine Ethics Committee, Kurdistan region, Iraq, on May 21, 2025 (Paper code: 13, Meeting:17). ResultsThirteen samples were tested for the presence of the Influenza A virus. Out of the 13, eight were positive (62%) for H5, and five (38%) samples gave negative results, as shown in Table 2. Out of 13 samples, nine (69%) were positive for the NA gene. Among these positive samples, eight (88%) were identified as N1 and one (11%) as N4, as shown in Table 2. The PCR amplification results of the HA gene, using specific primers designed for H5 genotype, are shown in Figure 4. These results confirmed that all eight positive samples carried the H5 genotype. The sequencing of the positive PCR product H5 genotype further confirmed the result. The result of annotating the sequenced samples using the NCBI-annotation tool revealed the H5 genotype, as shown in Figure 8. The HA gene for the KCVL15-013 sample was re-amplified to determine the exact size of the HA gene (1,771 bp), as shown in Figure 5. The PCR amplification of the NA, shown in Fig. 6, revealed positive results for the NA gene. After receiving the sequencing results from Genewiz and analyzing them using the annotation analysis (NCBI-annotation tool), one of the samples (KCVL15-013) was identified as an N4, and eight were identified as N1, as shown in Figures 9 and 10. This result was obtained through the accidental amplification of the N4 genotype during PCR amplification for the N1 genotype, using a set of primers initially designed for the N1 genotype, which resulted in a 600 bp PCR product (Fig. 6). A set of new specific primers was designed for N4 and used to re-amplify KCVL15-013, the sample KCVL15-013, for further confirmation. The result of re-amplifying KCVL15-013 showed a positive PCR product of 700 bp, corresponding to the N4 genotype (Fig. 7). The H5 and N4 results were confirmed by sequence annotation (Figs. 8 and 10). The results revealed the presence of the N4 genotype during the 2015 H5N1 pandemic in Iraq. By confirmation of H5 and N4 from the same source, our result revealed serotype H5N4. Table 2. Number of samples tested for HA and NA genotypes.
The H5 results were tested for pathogenicity. The Sanger sequencing and bioinformatics analysis of this HA gene cleavage site provided information, as it is a multibasic cleavage type RRRKR/GLF. The HPAI HA amino acid sequences of the cleavage site are mostly RRRKR/GLF; in contrast, the LPAI HA amino acid sequences of the cleavage site are EKQTR/GLF. Depending on these criteria, the pathogenicity type was confirmed, and the HA gene was identified as belonging to the HPAI H5 genotype, as shown in Fig. 11. The phylogenetic analysis of the N4 NA gene from influenza A viruses collected in 2015 showed that the strain KCVL15-013, detected in Iraq, formed a separate branch from all other sequences in the tree. It clustered only with another Iraqi chicken-origin strain (1735029.3|H5N4|Chicken|Iraq), with a low bootstrap support value of 37%. In contrast, the remaining sequences grouped into well-supported clades (100% bootstrap) according to geographic region and host species. Sequences from Blue-Winged Teal in Guatemala, Mallards and Northern Shovelers in the USA and Canada, and Ducks in Taiwan and China formed distinct clusters. These groupings reflect consistent clustering by host and location, whereas the two Iraqi chicken-origin strains, including KCVL15-013, fell outside the main clusters of wild birds, as shown in Fig. 12. DiscussionDuring the HPAI H5N1 outbreak in Iraq in 2015 (FAO, 2016), this study also detected the presence of the H5N4 serotype. Detecting H5N4 at the same time as H5N1 is important because it suggests that different strains of the virus may have been circulating together. This raises the possibility of reassortment between viruses, which could lead to the emergence of new, potentially highly pathogenic strains. It also has an impact on vaccination, as current vaccines are usually designed for specific strains, and the presence of multiple types could reduce their effectiveness (Tseng et al., 2024). This emphasizes the need for ongoing surveillance and flexible vaccination strategies (Iwami et al., 2009; Horwood et al., 2018; Parvin et al., 2018; Hew et al., 2024). As mentioned, the N4 gene in this study was accidentally amplified using N1-specific primers during molecular surveillance of HPAI H5N1. The cross-reactivity was likely due to partial sequence homology at the primer binding regions, allowing for the accidental amplification of an N4 gene. A similar remark was reported by Tsukamoto et al. (2009) during the validation of a conventional reverse transcription polymerase chain reaction method for subtyping influenza A NA genes (N1–N9). Although each primer set was designed to be subtype-specific, they found that occasional cross-reactivity occurred. In particular, some primer sets produced “faint, smeared, or size-different PCR products” when tested on non-target subtypes, and subsequent sequence analysis confirmed that these products were due to non-specific amplification or mixed subtype amplicons. This finding emphasizes the crucial importance of accompanying sequencing and annotation following PCR amplification, particularly in the molecular surveillance of influenza viruses. While PCR remains a commonly used and sensitive method for initial detection, it depends heavily on primer specificity, which may not always distinguish between closely related NA subtypes (Tsukamoto et al., 2009). Without follow-up sequencing, this cross-reactivity could lead to misidentification of subtypes, underreporting of rare variants, or inaccurate surveillance data. Sequencing of PCR products, followed by annotation using updated databases, provides complete subtype confirmation and enables the detection of unexpected or emerging strains (Alvarez et al., 2008). Multibasic amino acid cleavage sites are associated with increased disease severity in avian hosts, indicating a high level of virus replication and facilitating transmission (de Bruin et al., 2023). Detecting the amino acid cleavage site in the HA gene explains the virus’s virulence features (Runstadler et al., 2013; Nao et al., 2017). It was expected to identify the N4 to be related to the HPAI H5N4 strain, since the area had been invaded by the HPAI strain several times (Rashid et al., 2017; Saeed et al., 2021). This study helps us better understand how influenza A viruses can change and evolve. These viruses have eight-segmented genes, and when different strains infect the same host, they can mix and rearrange these segments in a process called reassortment (Webster et al., 1992; Nelson and Holmes, 2007). This can lead to the formation of new virus types with variable levels of severity or the ability to infect new hosts (Li et al., 2010). The appearance of H5N4 during the H5N1 outbreak suggests that genome reassortment may have occurred, especially in areas where multiple strains are circulating simultaneously (WHO, 2016). The study of this kind of genetic reassortment is important because it can lead to the emergence of new virulent viruses, making it harder to control outbreaks and maintain effective vaccination programs (Chen et al., 2006; Suarez, 2012). The phylogenetic location of the KCVL15-013 strain on the phylogenetic tree suggests that it may have evolved differently from other N4 influenza A viruses found in 2015. While most other viruses, especially those from wild birds in regions such as North America, Central America, and East Asia, grouped in well-supported clusters, KCVL15-013 grouped only with one other Iraqi chicken-origin strain. It showed weak bootstrap support (37%), suggesting limited genetic similarity, even within the same host and country. This divergence may reflect localized virus evolution, potentially driven by different ecological pressures, biosecurity practices, or host dynamics in poultry farming environments. The lack of closely related sequences from surrounding regions further emphasizes the gap in influenza surveillance in the Middle East, which limits our understanding of viral diversity and transmission pathways. These findings underscore the importance of enhancing genomic surveillance in poultry, particularly in underrepresented areas, to detect emerging variants early and assess their potential impact on animal and public health. ConclusionVeterinary diagnostic molecular laboratories worldwide should consider that other serotypes may cause HPAI in combination with the common serotype. Developing a poultry vaccine program, tailored to the specific serotype, is necessary to combat AI infection. Surveillance and epidemiological studies in different avian species must start to identify the incidence of AI disease. Future research should aim to better understand how new strains of avian influenza are evolving. It’s also important to test how well current vaccines, especially those designed to protect against multiple strains, work against these emerging threats. Additionally, understanding how the virus mixes its genes (reassortment) and how it transmits between different animal species will be crucial. These efforts will help us stay better prepared for future outbreaks and guide the development of more effective prevention and control strategies. AcknowledgmentsThe authors would like to thank all those who helped to achieve this research. Conflict of interestThe authors declare that they have no competing interests. FundingThis study has not been funded by any research grant. Authors’ contributionsB.A.D. provided the experiment requirements (materials, machine, and lab space). E.L.K. conceived the design, conducted the experiments, and wrote the manuscript. Data availabilityThe datasets used and analyzed during the current study are available from the corresponding author upon reasonable request. ReferencesAlexander, D.J. 2007. An overview of the epidemiology of avian influenza. Vaccine 25(30), 5637–5644; doi:10.1016/j.vaccine.2006.10.051 Al-Ouqaili, M.T.S., Majeed, Y.H. and Al-Ani, S.K. 2020. SEN virus genotype H distribution in β-thalassemic patients and in healthy donors in Iraq: molecular and physiological study. PLos Neglected. Trop. Dis. 14(6), 7880. Alvarez, A.C., Brunck, M.E., Boyd, V., Lai, R., Virtue, E., Chen, W., Bletchly, C., Heine, H.G. and Barnard, R. 2008. A broad spectrum, one-step reverse-transcription PCR amplification of the neuraminidase gene from multiple subtypes of influenza A virus. Virol. J. 5(1), 77; doi:10.1186/1743-422X-5-77 Arbeitskreis Blut, U.B.B.K. 2009. Influenza Virus. Transfus. Med. Hemother. 36(1), 32–39; doi:10.1159/000197314 Brown, J.D., Stallknecht, D.E., Berghaus, R.D. and Swayne, D.E. 2009. Infectious and lethal doses of H5N1 highly pathogenic avian influenza virus for house sparrows (Passer domesticus) and rock pigeons (Columbia livia). J. Vet. Diagnostic Invest. 21(4), 437–445; doi:10.1177/104063870902100404 Capua, I. and Alexander, D.J. 2004. Avian influenza: recent developments. Avian. Pathol. 33(4), 393–404; doi:10.1080/03079450410001724085 Chen, H., Smith, G.J.D., Li, K.S., Wang, J., Fan, X.H., Rayner, J.M., Vijaykrishna, D., Zhang, J.X., Zhang, L.J., Guo, C.T., Cheung, C.L., Xu, K.M., Duan, L., Huang, K., Qin, K., Leung, Y.H.C., Wu, W.L., Lu, H.R., Chen, Y., Xia, N.S., Naipospos, T.S.P., Yuen, K.Y., Hassan, S.S., Bahri, S., Nguyen, T.D., Webster, R.G., Peiris, J.S.M. and Guan, Y. 2006. Establishment of multiple sublineages of H5N1 influenza virus in Asia: implications for pandemic control. Proc. Nat. Acad. Sci. 103(8), 2845–2850. CIDRAP. 2006. Iraq confirms human bird flu case, suspects more. Available via https://www.cidrap.umn.edu/avian-influenza-bird-flu/iraq-confirms-human-bird-flu-case-suspects-more?utm_source=chatgpt.com Dadonaite, B., Gilbertson, B., Knight, M.L., Trifkovic, S., Rockman, S., Laederach, A., Brown, L.E., Fodor, E. and Bauer, D.L.V. 2019. The structure of the influenza A virus genome. Nature. Microbiol. 4(11), 1781–1789; doi:10.1038/s41564-019-0513-7 De Bruin, A.C.M., Spronken, M.I., Bestebroer, T.M., Fouchier, R.A.M. and Richard, M. 2023. Conserved expression and functionality of Furin between chickens and ducks as an activating protease of highly pathogenic avian influenza virus hemagglutinins. Microbiol. Spectr. 11(2), e04602–e04622; doi:10.1128/spectrum.04602-2 De Graaf, M. and Fouchier, R.A.M. 2014. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO. J. 33(8), 823–841; doi:10.1002/embj.201387442 Dugan, V.G., Chen, R., Spiro, D.J., Sengamalay, N., Zaborsky, J., Ghedin, E., Nolting, J., Swayne, D.E., Runstadler, J.A., Happ, G.M., Senne, D.A., Wang, R., Slemons, R.D., Holmes, E.C. and Taubenberger, J.K. 2008. The evolutionary genetics and emergence of avian influenza viruses in wild birds. PLos Pathogens. 4(5), e1000076; doi:10.1371/journal.ppat.1000076 FAO. 2022. Influenza A Cleavage Sites. OIE & FAO. Available via https://www.offlu.org/wp-content/uploads/2022/01/Influenza-A-Cleavage-Sites-Final-04-01-2022.pdf Ghedin, E., Sengamalay, N.A., Shumway, M., Zaborsky, J., Feldblyum, T., Subbu, V., Spiro, D.J., Sitz, J., Koo, H., Bolotov, P., Dernovoy, D., Tatusova, T., Bao, Y., St George, K., Taylor, J., Lipman, D.J., Fraser, C.M., Taubenberger, J.K. and Salzberg, S.L. 2005. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature 437(7062), 1162–1166; doi:10.1038/nature04239 Hew, Y.L., Hiono, T., Monne, I., Nabeshima, K., Sakuma, S., Kumagai, A., Okamura, S., Soda, K., Ito, H., Esaki, M., Okuya, K., Ozawa, M., Yabuta, T., Takakuwa, H., Nguyen, L.B., Isoda, N., Miyazawa, K., Onuma, M. and Sakoda, Y. 2024. Cocirculation of genetically distinct highly pathogenic avian influenza H5N5 and H5N1 viruses in crows, Hokkaido, Japan. Emerg. Infect. Dis. 30(9), 1912. Horwood, P.F., Horm, S.V., Suttie, A., Thet, S., Y, P., Rith, S., Sorn, S., Holl, D., Tum, S., Ly, S., Karlsson, E.A., Tarantola, A. and Dussart, P. 2018. Co-circulation of influenza A H5, H7, and H9 viruses and co-infected poultry in live bird markets, Cambodia. Emerg. Infect. Dis. 24(2), 352. Iwami, S., Suzuki, T. and Takeuchi, Y. 2009. Paradox of vaccination: is vaccination really effective against avian flu epidemics?. PLos One 4(3), e4915; doi:10.1371/journal.pone.0004915 King, J., Harder, T., Globig, A., Stacker, L., Günther, A., Grund, C., Beer, M. and Pohlmann, A. 2022. Highly pathogenic avian influenza virus incursions of subtype H5N8, H5N5, H5N1, H5N4, and H5N3 in Germany during 2020-21. Virus. Evol. 8(1), 35; doi:10.1093/ve/veac035 Li, C., Hatta, M., Nidom, C.A., Muramoto, Y., Watanabe, S., Neumann, G. and Kawaoka, Y. 2010. Reassortment between avian H5N1 and human H3N2 influenza viruses creates hybrid viruses with substantial virulence. Proc. Nat. Acad. Sci. 107(10), 4687–4692. Nao, N., Yamagishi, J., Miyamoto, H., Igarashi, M., Manzoor, R., Ohnuma, A., Tsuda, Y., Furuyama, W., Shigeno, A., Kajihara, M., Kishida, N., Yoshida, R. and Takada, A. 2017. Genetic predisposition to acquire a polybasic cleavage site for highly pathogenic avian influenza virus hemagglutinin. MBio 8(1), e02298-16. doi: 10.1128/mbio.02298-16 Nelson, M.I. and Holmes, E.C. 2007. The evolution of epidemic influenza. Nature Rev. Genet. 8(3), 196–205; doi:10.1038/nrg2053 Neumann, G., Shinya, K. and Kawaoka, Y. 2007. Molecular pathogenesis of H5N1 influenza virus infections. Antiviral. Therapy. 12(4_part_2), 617–626; doi:10.1177/135965350701200S08.1 OIE. 2018. World Organization for Animal Health (OIE). OIE Situation Report for avian influenza. Available via https://www.woah.org/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/OIE_AI_situation_report/OIE_SituationReport_AI_June2018.pdf Parvin, R., Begum, J.A., Nooruzzaman, M., Chowdhury, E.H., Islam, M.R. and Vahlenkamp, T.W. 2018. Review analysis and impact of co-circulating H5N1 and H9N2 avian influenza viruses in Bangladesh. Epidemiol. Infect. 146(10), 1259–1266; doi:10.1017/s0950268818001292 Rashid, P.M.A., Saeed, N.M. and Dyary, H.O. 2017. Genetic characterization and phylogenic analysis of H5N1 avian influenza virus detected in peafowl in Kirkuk province, Iraq. J. Med. Virol. 89(7), 1179–1185; doi:10.1002/jmv.24762 Runstadler, J., Hill, N., Hussein, I.T.M., Puryear, W. and Keogh, M. 2013. Connecting the study of wild influenza with the potential for pandemic disease. Infection. Genet. Evol. 17, 162–187; doi:10.1016/j.meegid.2013.02.020 Saeed, N.M., Rashid, P.M.A. and Dyary, H.O. 2021. Genetic characterization of highly pathogenic avian influenza A (H5N8) virus isolated from domestic geese in Iraq, 2018. BMC. Vet. Res. 17, 1–7; doi:10.1186/s12917-021-02831-y Sambrook, J. 1989. Molecular cloning: a laboratory. New York, NY: Cold Spring Harbor Laboratory. Senne, D.A., Panigrahy, B., Kawaoka, Y., Pearson, J.E., Suss, J., Lipkind, M., Kida, H. and Webster, R.G. 1996. Survey of the hemagglutinin (HA) cleavage site sequence of H5 and H7 avian influenza viruses: amino acid sequence at the HA cleavage site as a marker of pathogenicity potential. Avian Dis. 40, 425–437; doi: 10.2307/1592241 Suarez, D.L. 2000. Evolution of avian influenza viruses. Vet. Microbiol. 74(1-2), 15–27; doi:10.1016/S0378-1135(00)00163-4 Suarez, D.L. 2012. DIVA vaccination strategies for avian influenza virus. Avian. Dis. 56(4s1), 836–844. Sudhir Kumar, G.S., Suleski, M., Sanderford, M., Sharma, S. and Tamura, K. 2024. MEGA12: molecular evolutionary genetic analysis version for adaptive and green computing. Mol. Biol. Evol. 41(12), 1–9; doi: 10.1093/molbev/msae263 Tamura, K. and Nei, M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10(3), 512–526; doi:10.1093/oxfordjournals.molbev.a040023 Tseng, I., Pan, B.Y., Feng, Y.C. and Fang, C.T. 2024. Re-evaluating efficacy of vaccines against highly pathogenic avian influenza virus in poultry: a systematic review and meta-analysis. One Health 18, 100714; doi:10.1016/j.onehlt.2024.100714 Tsukamoto, K., Ashizawa, T., Nakanishi, K., Kaji, N., Suzuki, K., Shishido, M., Okamatsu, M. and Mase, M. 2009. Use of reverse transcriptase PCR to subtype N1 to N9 neuraminidase genes of avian influenza viruses. J. Clin. Microbiol. 47(7), 2301–2303; doi:10.1128/jcm.02366-08 Venkatesh, D., Poen, M.J., Bestebroer, T.M., Scheuer, R.D., Vuong, O., Chkhaidze, M., Machablishvili, A., Mamuchadze, J., Ninua, L. and Fedorova, N.B. 2018a. Avian influenza viruses in wild birds: virus evolution in a multihost ecosystem. J. Virol. 92(15), e00433-18. doi: 10.1128/jvi.00433-18 Webster, R.G., Bean, W.J., Gorman, O.T., Chambers, T.M. and Kawaoka, Y. 1992. Evolution and ecology of influenza A viruses. Microbiol. Rev. 56(1), 152–179; doi:10.1128/mr.56.1.152-179.1992 WHO. 2016. Antigenic and genetic characteristics of zoonotic influenza A viruses and development of candidate vaccine viruses for pandemic preparedness. WOAH/OIE. 2023. Avian Influenza. Available via https://www.woah.org/en/disease/avian-influenza/ Xie, Z., Yang, J., Jiao, W., Li, X., Iqbal, M., Liao, M. and Dai, M. 2025. Clade 2.3.4.4b highly pathogenic avian influenza H5N1 viruses: knowns, unknowns, and challenges. J. Virol. 99(6), 42425; doi:10.1128/jvi.00424-25 | ||








| How to Cite this Article |
| Pubmed Style Dizayee BA, Khlef EL. Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak. Open Vet. J.. 2025; 15(10): 5312-5325. doi:10.5455/OVJ.2025.v15.i10.48 Web Style Dizayee BA, Khlef EL. Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak. https://www.openveterinaryjournal.com/?mno=252729 [Access: January 25, 2026]. doi:10.5455/OVJ.2025.v15.i10.48 AMA (American Medical Association) Style Dizayee BA, Khlef EL. Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak. Open Vet. J.. 2025; 15(10): 5312-5325. doi:10.5455/OVJ.2025.v15.i10.48 Vancouver/ICMJE Style Dizayee BA, Khlef EL. Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak. Open Vet. J.. (2025), [cited January 25, 2026]; 15(10): 5312-5325. doi:10.5455/OVJ.2025.v15.i10.48 Harvard Style Dizayee, B. A. & Khlef, . E. L. (2025) Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak. Open Vet. J., 15 (10), 5312-5325. doi:10.5455/OVJ.2025.v15.i10.48 Turabian Style Dizayee, Bejan Ahmad, and Evan Latef Khlef. 2025. Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak. Open Veterinary Journal, 15 (10), 5312-5325. doi:10.5455/OVJ.2025.v15.i10.48 Chicago Style Dizayee, Bejan Ahmad, and Evan Latef Khlef. "Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak." Open Veterinary Journal 15 (2025), 5312-5325. doi:10.5455/OVJ.2025.v15.i10.48 MLA (The Modern Language Association) Style Dizayee, Bejan Ahmad, and Evan Latef Khlef. "Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak." Open Veterinary Journal 15.10 (2025), 5312-5325. Print. doi:10.5455/OVJ.2025.v15.i10.48 APA (American Psychological Association) Style Dizayee, B. A. & Khlef, . E. L. (2025) Molecular characterization of a highly pathogenic avian influenza A (H5N4) virus isolated from domestic chickens in Iraq during the 2015 HPAI H5N1 outbreak. Open Veterinary Journal, 15 (10), 5312-5325. doi:10.5455/OVJ.2025.v15.i10.48 |

