




| Research Article | ||
Open Vet. J.. 2025; 15(10): 5183-5191 Open Veterinary Journal, (2025), Vol. 15(10): 5183-5191 Research Article Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategyHanwen Hu1 and Wei Xie2*1Central Bucks High School East, Doylestown, PA, USA 2MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory for Biocontrol, School of Life Sciences, Sun Yat-Sen University, Guangzhou, People’s Republic of China *Corresponding Author: Wei Xie. MOE Key Laboratory of Gene Function and Regulation, State Key Laboratory for Biocontrol, School of Life Sciences, Sun Yat-Sen University, Guangzhou, People’s Republic of China. Email: xiewei6 [at] mail.sysu.edu.cn Submitted: 01/07/2025 Revised: 21/09/2025 Accepted: 29/09/2025 Published: 31/10/2025 © 2025 Open Veterinary Journal
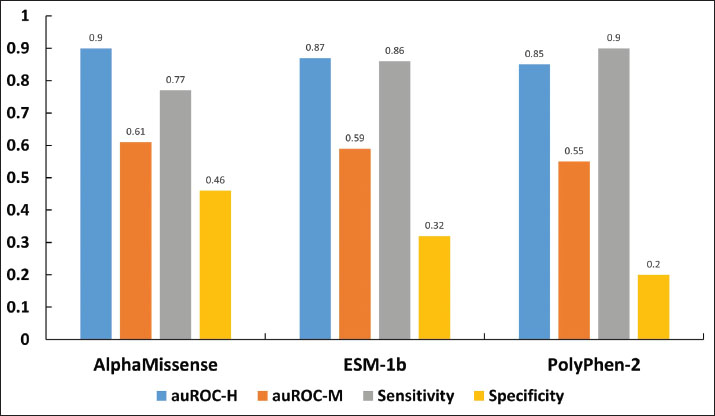
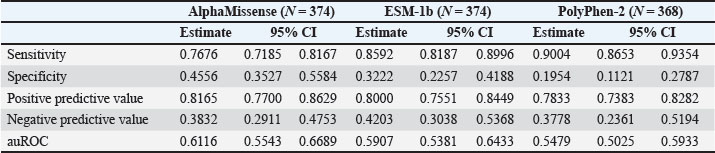
AbstractBackground: Predicting the pathogenicity of missense variants is a crucial step in animal breeding, farming, and veterinary medicine. While some predictive tools for non-human species exist, many are not as specialized or effective as those developed for humans. Existing human genomic tools can provide insights that may be beneficial when appropriately adapted for animal use due to protein sequence similarities between humans and other mammals. Aim: This study aimed to evaluate the applicability of three widely used human genomic prediction tools—AlphaMissense, ESM-1b, and PolyPhen-2—to assess the pathogenicity of missense variants in domestic mammals. Methods: Missense variants in domestic mammals were sourced from the Online Mendelian Inheritance in Animals database, comprising 284 variants annotated as “deleterious” and 90 as “non-deleterious.” These variants were converted into mock human variants and classified as pathogenic or benign using the three human genomic tools. Predictive performance was assessed by calculating the sensitivity, specificity, and area under the receiver operating characteristic curve (auROC). Results: The tools showed sensitivity values between 0.77 and 0.90, indicating a strong ability to identify pathogenic variants, but low specificity values from 0.20 to 0.46 suggest challenges in correctly identifying benign variants, with auROC values indicating moderate predictive performance ranging from 0.55 to 0.61. The low specificity may stem from the interpretation of “non-deleterious” variants, which may cause malfunctioning proteins that are often associated with traits such as coat color but are not linked to disease in animals. Conclusion: A simple mock strategy can enable human genomic tools to predict the pathogenicity of rare missense variants in domestic mammals. Although specificity remains a limitation, and the strategy is primarily applicable to mutations on highly conserved amino acid residues, the results highlight the applicability for adapting human genomic prediction tools to advance veterinary genomics and animal research, with predictive performance potentially influenced by the sequence similarity between animal and human homologous proteins. Keywords: Genomic prediction tools, Pathogenicity, Missense variants, Animals. IntroductionRecent advances in gene sequencing technologies have significantly enhanced the understanding of specific aspects of animal genomics, enabling detailed studies of genotypes and diseases in livestock and companion animals. The identification of certain single-nucleotide polymorphisms as genetic markers has improved breeding value predictions and disease-associated variant detection, propelling animal research into a new era of precision science (Rexroad et al., 2019). Unlike deletion and nonsense mutations, missense variants can have diverse effects that complicate their interpretation. While numerous genomic tools have been developed to predict the disease risk of human missense variants, many tools for animal genomics are not as developed as those for humans, indicating a need for further research and innovation (Yang et al., 2022). Building on findings from recent research that compared rare missense variants and disease associations between animals and humans to enrich human studies (Haque et al., 2024), we hypothesized that human genomic tools could be simply adopted as an effective method for interpreting the pathogenicity of mammalian variants by employing mock variants generated by mapping mammalian variants onto homologous human protein sequences. To test this hypothesis, we evaluated the performance of three widely used human genomic prediction tools—AlphaMissense (Cheng et al., 2023), ESM-1b (Brandes et al., 2023), and PolyPhen-2 (Adzhubei et al., 2010)—on missense variants from domestic mammals documented in the Online Mendelian Inheritance in Animals (OMIA) database (Nicholas et al., 2025), which contains 1,636 genetic variants across 602 species, with missense variants being the most common as of 2024. The resulting high sensitivity, coupled with the seemingly counterintuitive low specificity, underscores the practical utility of the mock strategy in adapting human genomic tools to predict the pathogenicity of missense variants in animals. Materials and MethodsObtaining variants of domestic mammals and generating mock human equivalentsMissense variant data specifically related to eight domestic mammals, dog, cattle, cat, horse, sheep, pig, goat, and rabbit, were extracted from the OMIA dataset. The raw OMIA data underwent a rigorous manual verification process to ensure data quality, which involved validating annotations, addressing missing entries, and standardizing formatting inconsistencies through established protocols. For each validated missense variant, corresponding protein information for both the animal and its human homologs—including UniProt IDs, entry names, NCBI IDs, and sequences—was retrieved from the UniProt and NCBI databases using Python scripts. The animal missense variants were aligned and mapped to corresponding residues in the human homologous protein sequence using sequence alignment tools of the BioPython package with default settings (Cock et al., 2009), then the animal missense variants were mapped to the corresponding residues in the human homologous protein sequence to generate the mock human equivalents. Thus, mock human missense variants were generated. We discarded animal variants for which no corresponding residues could be identified in the human homologous protein. Pathogenicity of missense variantsOMIA variant classification was assigned binary labels as “deleterious” to 1 (pathogenic) and “non-deleterious” to 0 (benign). The AlphaMissense, ESM-1b, and PolyPhen-2 prediction outputs wereprediction outputs of AlphaMissense, ESM-1b, and PolyPhen-2 were processed and classified according to the following criteria: AlphaMissense: The mock human missense variants were assigned a label of 1 if they were classified as “Lpath” (likely pathogenic) and a label of 0 if they were classified as “Lben” (likely benign) or “Amb” (ambiguous). ESM-1b: Variants were classified as 1 if the prediction score was less than −7.5, and as 0 if the score was greater than or equal to −7.5 (Goldman et al. 2023) (Goldman et al. 2023). PolyPhen-2: Variants were labeled 1 if classified as “probably damaging” or “possibly damaging” and 0 if classified as “benign.” Statistical methodsThe performance of these human genomic tools in classifying the pathogenicity of missense variants was evaluated based on their prediction accuracy. The sensitivity, specificity, positive predictive values, negative predictive values, and area under the receiver operating characteristic curve (auROC) were reported as point estimates and 95% confidence intervals (CIs). Logistic regression models were used to assess the impact of species on predicting the pathogenicity of missense variants in domestic mammals for the three tools separately. The pathogenicity status (based on each tool) and the species category (with the species having the largest sample size used as the reference group) were included as independent variables in these models, while the OMIA variant classification served as the dependent variable. Statistical analyses, including adjusted analyses, were performed using the SAS® OnDemand for Academics software. ResultsA total of 374 missense variants were employed for the generation of mock human variants, pathogenicity prediction, and further analysis. They were distributed across eight species as follows: 126 in dogs, 93 in cattle, 60 variants in cats, 43 in horses, 29 in sheep, 15 in pigs, 5 in rabbits, and 3 in goats. The variants spanned 236 distinct genes, with 176 genes harboring only a single variant. The MC1R and KIT genes exhibited the highest numbers of variants, with 19 and 13 variants, respectively. Table S1 of Supplementary Data summarizes the 374 missense variants, their corresponding mock human variants, and the prediction results from AlphaMissense, ESM-1b, and PolyPhen-2. All three tools exhibited high sensitivity and low specificityAmong these 374 animal missense variants, 284 (76%) were annotated as “deleterious,” whereas 90 (24%) were annotated as “non-deleterious” by the OMIA dataset. For their mock human variants, AlphaMissense identified 267 variants (71.4%) as pathogenic and 107 (28.6%) as benign; ESM-1b predicted 305 (81.6%) as pathogenic and 69 (18.5%) as benign; PolyPhen-2 classified 323 (86.4%) as pathogenic, 45 (12.0%) as benign, and six variants (1.6%) were missed. Benchmark tests using three human variant datasets—ClinVar (class-balanced, 18,924 variants), ClinVar (612 genes), and DDD (410 de novo variants)—revealed that AlphaMissense achieved the highest accuracy, with auROC values of 0.93, 0.95, and 0.81, respectively (Cheng et al., 2023). In comparison, ESM-1b attained auROC values of 0.92, 0.91, and 0.78, whereas PolyPhen-2 yielded values of 0.88, 0.89, and 0.77 (Cheng et al., 2023). With OMIA dataset classification of these 374 domestic animal variants as the gold standard, the three tools exhibited sensitivity values ranging from 0.77 to 0.90 (a sensitivity of one means that all actual positive cases are identified as positive by prediction): AlphaMissense at 0.77 (95% CI: 0.72–0.82), ESM-1b at 0.86 (95% CI: 0.82–0.90), and PolyPhen-2 at 0.90 (95% CI: 0.87–0.94), highlighting their strong performance in detecting “deleterious” (pathogenic) variants. However, their specificity values ranged from 0.20 to 0.46 (a specificity of one means that all actual negative cases are identified as negative by prediction), with values of 0.46 (95% CI: 0.35–0.56) for AlphaMissense, 0.32 (95% CI: 0.23–0.42) for ESM-1b, and 0.20 (95% CI: 0.11–0.28) for PolyPhen-2, highlighting their poor ability to identify “non-deleterious” (benign) variants. Therefore, all three tools showed moderate accuracy in predicting the pathogenicity of these variants. AlphaMissense achieved the highest accuracy at 0.61 (95% CI: 0.55–0.67), followed by ESM-1b at 0.59 (95% CI: 0.54–0.64) and PolyPhen-2 at 0.55 (95% CI: 0.50–0.59) (Figs. 1 and S1). Additionally, the positive and negative predictive values are detailed in the supplementary materials (Table S2).
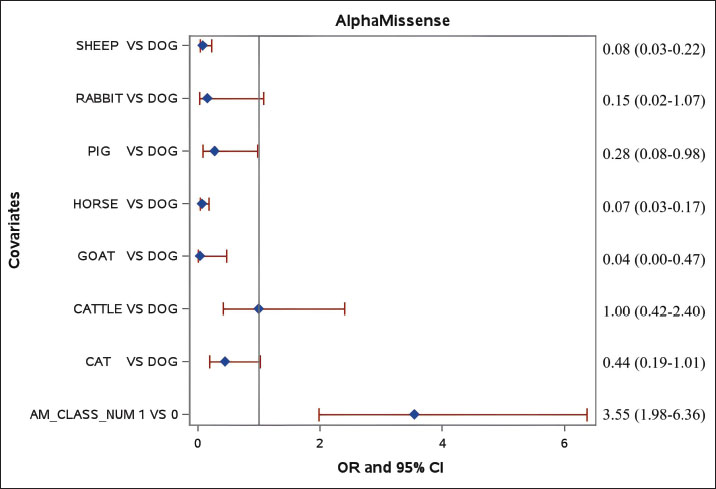
Fig. 1. Performance of AlphaMissense, ESM-1b, and PolyPhen-2 in predicting the pathogenicity of 374 domestic animal variants using the mock strategy. auROC-H: average auROC for three human variant datasets [4], auROC-M=average auROC for mock human variants Species significantly associated with the prediction performanceAdjusted analysis was conducted to identify factors influencing the prediction performance of the three tools. For AlphaMissense, compared with the reference group (dogs), goats (odds ratio [OR]=0.04, 95% CI: 0.00–0.47, p=0.0107), horses (OR=0.07, 95% CI: 0.03–0.17, p < 0.0001), sheep (OR=0.08, 95% CI: 0.03–0.22, p < 0.0001), and pigs (OR=0.28, 95% CI: 0.08–0.98, p=0.0458) exhibited significantly lower odds of being classified as pathogenic, with CIs excluding one, indicating robust differences. In contrast, cats (OR=0.44, 95% CI: 0.19–1.01, p=0.0540), cattle (OR=1.00, 95% CI: 0.42–2.40, p=0.9981), and rabbits (OR=0.15, 95% CI: 0.02–1.07, p=0.0585) showed no significant difference from dogs, as their CIs included one. The results suggest that species-specific genetic characteristics or imbalances in training data—grouped by species—play a critical role in prediction outcomes (Fig. 2). The low ORs observed for horses align with similar trends in goats and sheep, indicating a broader pattern rather than an extreme horse-specific bias. However, this consistency raises the possibility of underrepresentation of pathogenic mutations in the training data for these species, which may contribute to reduced accuracy for animal variant prediction. These findings highlight the importance of further investigating species-specific data for the performance of AlphaMissense. Parallel analyses using ESM-1b and PolyPhen-2 yielded comparable results, with their supporting data detailed in the supplementary materials (Fig. S2A and B).
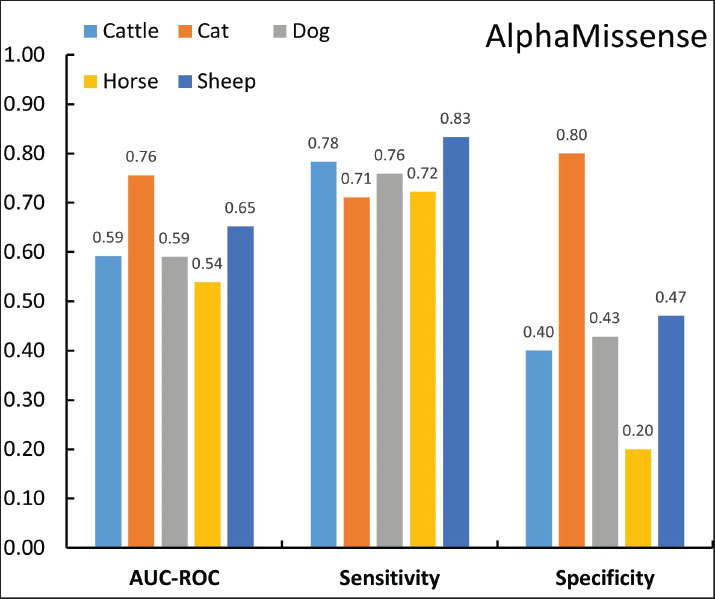
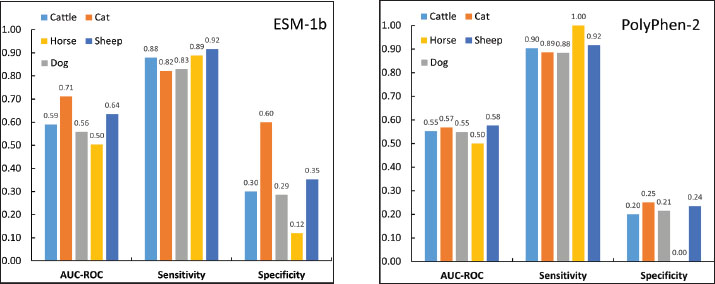
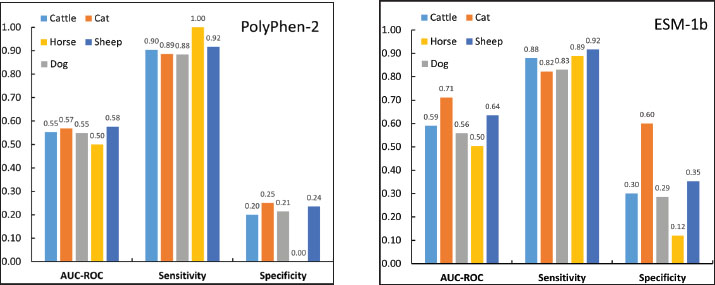
Fig. 2. Species effects on pathogenicity prediction of missense variants in domestic mammals (logistic regression, n=374, AlphaMissense). AM-CLASS_NUM=predicted pathogenicity status. Subsequent analyses of prediction performance were conducted separately for data from cats, cattle, dogs, sheep, and horses. Data from goats, pigs, and rabbits were excluded due to limited sample sizes. AlphaMissense performance varies significantly across species, with auROC values reaching their highest of 0.76 (95% CI: 0.63–0.88) with cat data and dropping to 0.54 (95% CI: 0.41–0.67) with horse data. Sensitivities remain consistent across all five species, ranging from 0.71 (95% CI: 0.58–0.84) for cats to 0.83 (95% CI: 0.62–1.00) for sheep, suggesting that differences in accuracy are primarily driven by variations in specificity, which is 0.80 (95% CI: 0.60–1.00) for cats and 0.20 (95% CI: 0.04–0.36) for horses (Fig. 3). In contrast, PolyPhen-2 shows consistently low auROC values ranging from 0.50 (95% CI: 0.50–0.50) for horses to 0.58 (95% CI: 0.44–0.71) for sheep across the five species. While its sensitivities are high, ranging from 0.88 (95% CI: 0.82–0.94) for dogs to 1.00 (95% CI: 0.79–1.00) for horses, its specificities are uniformly low (from 0.00 [95% CI: 0.00%–0.14%] for horses to 0.25 [95% CI: 0.01–0.50] for cats) for all species (Fig. S3A). The performance of ESM-1b is comparable to that of AlphaMissense (Fig. S3B).
Fig. 3. Species-specific performance of AlphaMissense: sensitivity, specificity, and auROC values for cattle, cats, dogs, horses, and sheep. Low specificity: non-deleterious but pathogenicAt least one tool predicted the pathogenicity of 284 animal missense variants labeled as “deleterious” in OMIA. Cross-referencing with OMIA data revealed that these variants are predominantly associated with severe or lethal diseases, such as bulldog calf syndrome, myotonia, and osteogenesis imperfecta (Amills, 2025). In contrast, for the 90 variants classified as “non-deleterious” in OMIA, which are mostly associated with traits such as coat color and hair texture or linked to reproductive traits such as litter size, 59 were predicted to be pathogenic by at least two tools, and 45 were predicted to be pathogenic by all three tools. Notably, 23 of these 59 variants, 23 originate from the horse data, which includes only 43 variants. Thus, a simple explanation may exist for the observed low specificity. Some variants significantly interfere with the normal physiological functions of their associated proteins and are correctly predicted as "pathogenic" by the tools. However, they are labeled as "non-deleterious" in OMIA because the resulting traits in animals do not impair normal functionality and are often considered desirable new characteristics. For example, the horse KIT gene variants p. G597R, p. A602V, p. G654R, p. L674H, p. L674P, p. E667D, p. N783Y, and p. K830I are labeled as "non-deleterious" in OMIA and are predicted as "pathogenic" by the tools. These variants were associated with depigmentation of the skin and hair in horses, leading to desirable traits such as a white coat or white spots (Haase et al., 2007, 2009, 2011; Hauswirth et al., 2013), which is apparently due to the inability of the KIT gene product, tyrosine kinase, to properly produce melanin in the presence of these variants (Pham et al., 2020). In humans, the same depigmentation, manifested as a condition known as piebaldism, is classified as a disease (Spritz et al., 1992). Evidently, had these mutations been discovered in humans, they would likely be annotated as “pathogenic” in human mutation databases like ClinVar, thus rendering these tools’ predictions accurate. An intriguing example of this observation comes from four mutations in the MC1R gene of pigs, which also demonstrates the study’s potential practical applications. Black coat color in pigs is determined by the dominant E allele at the MC1R locus, and four missense variants in the MC1R gene—p. A243T, p. A164V, p. L102P, and p. V95M—are believed to play a role in eumelanin synthesis and are labeled as "non-deleterious" in the OMIA database. Researchers successfully transformed red-skinned Duroc pigs into black ones by replacing all four missense variants (Zhong et al., 2022). However, this study revealed that only one of them, p. L102P, is highly likely pathogenic. Editing this single variant might achieve the same result, significantly simplifying the procedure and reducing costs. ConclusionsThis study demonstrates the applicability of adapting human genomic tools—namely AlphaMissense, ESM-1b, and PolyPhen-2—to predict the pathogenicity of rare missense variants in animals cataloged in the OMIA database, employing a straightforward mock human variant strategy. Compared with existing species-specific predictors, such as PON-All, an important advantage of our approach is the avoidance of data circularity, as the OMIA database already incorporates the training dataset of PON-All. These tools demonstrated high sensitivity (0.77–0.90) when evaluated on 374 variants from eight domestic mammals, indicating their excellent capability to identify deleterious variants that are frequently associated with severe or lethal phenotypes that disrupt protein function. However, the tools exhibited low specificity (0.20–0.46) for “non-deleterious” variants, resulting in a moderate overall predictive performance, with auROC values ranging only from 0.55 to 0.61. At first glance, this mock strategy may appear to have limited practical application. However, a closer examination of the OMIA dataset reveals a critical nuance. The low specificity may arise from discrepancies in how “non-deleterious” variants are classified in OMIA compared to the “benign” used by the genomic tools, even though “deleterious” variants can be directly equated with “pathogenic.” Many variants listed as “non-deleterious” in OMIA, particularly those associated with traits such as coat color, were actually predicted as pathogenic by the three tools. This is because these variants do alter protein function; they are only labeled “non-deleterious” because the resulting physical characteristics are considered desirable traits rather than diseases. Consequently, only some of these “non-deleterious” variants are truly benign. Take the four “non-deleterious” MC1R variants in pigs (p. A243T, p. A164V, p. L102P, and p. V95M) as an example. It is highly probable that only p. L102P is “pathogenic.” This specific mutation causes the MC1R protein to lose its normal function, leading to a lack of eumelanin, leading to white or red pig skin. However, the other three mutations are truly benign, neither disrupting MC1R protein function nor influencing the coat color of the pig. Far from a failure, the lower specificity and moderate predictive performance we observed reveal a fundamental difference in definitions: the OMIA database’s classification of “non-deleterious” variants diverges from the “benign” designation used by genomic tools. These findings highlight the importance of refining annotations in animal variant databases. Moving forward, it is essential to clearly distinguish between truly benign variants and those that alter protein function, whether they lead to non-disease traits or to actual diseases. The small size of the OMIA dataset may introduce prediction biases, particularly for “non-deleterious” variants; further expansion and refinement of the OMIA dataset could enhance robustness. Additionally, as a preliminary attempt, this study used only three representative human genomic tools. Testing a broader range of tools could yield varied prediction outcomes, potentially improving animal mutation prediction accuracy. Although this mock variant approach can theoretically be applied to missense mutations in species evolutionarily close to humans, such as mammals, as demonstrated in our results, the mock strategy was exclusively applied to mutations located within highly conserved amino acid residues. It is crucial to acknowledge that a direct human mock residue correspondence does not exist for every animal protein residue; in fact, some may have no human equivalent, thus invalidating this mock approach for such instances. Moreover, our preliminary data indicate that sequence similarity between animal and human homologous proteins has no apparent significant effect on predictive performance. We are currently undertaking additional investigations to identify factors that may influence predictive performance. AcknowledgmentThe authors would like to thank the Pennsylvania Junior Academy of Science and Delaware Valley Science Fair for recognizing this research. Conflicts of interestThe authors have no competing interests to declare. FundingNo funding to declare. Authors’ contributionsWei Xie and Hanwen Hu: Conceptualization and design. Hanwen Hu: Data collection and analysis. Wei Xie and Hanwen Hu: Writing, editing, and reviewing the manuscript. Ethical approvalThis study did not require ethical approval because it involved no experiments on live animals or humans. Data availabilityAll data are included in the supplementary materials, and the code used in this study is available upon request from the corresponding author or from https://github.com/xhu001/OMIA-Prediction. ReferencesAdzhubei, I.A., Schmidt, S., Peshkin, L., Ramensky, V.E., Gerasimova, A., Bork, P., Kondrashov, A.S. and Sunyaev, S.R. 2010. A method and server for predicting damaging missense mutations. Nat. Methods. 7(4), 248–249. Amills, M. 2025. Cataloguing deleterious variants in domestic animal species: motivation, applications and challenges. Livestock Sci. 105721. Brandes, N., Goldman, G., Wang, C.H., Ye, C.J. and Ntranos, V. 2023. Genome-wide prediction of disease variant effects with a deep protein language model. Nat. Genet. 55(9), 1512–1522. Cheng, J., Novati, G., Pan, J., Bycroft, C., Žemgulytė, A., Applebaum, T., Pritzel, A., Wong, L.H., Zielinski, M., Sargeant, T., Schneider, R.G., Senior, A.W., Jumper, J., Hassabis, D., Kohli, P. and Avsec, Z. 2023. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 381(6664), eadg7492. Cock, P.J.A., Antao, T., Chang, J.T., Chapman, B.A., Cox, C.J., Dalke, A., Friedberg, I., Hamelryck, T., Kauff, F., Wilczynski, B. and De Hoon, M.J.L. 2009. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25(11), 1422. Haase, B., Brooks, S.A., Tozaki, T. and Burger, D. 2009. Seven novel KIT mutations in horses with white coat colour phenotypes. Anim. Genet. 40(5), 623–629. Haase, B., Rieder, S., Tozaki, T., Hasegawa, T., Penedo, M.C.T., Jude, R. and Leeb, T. 2011. Five novel KIT mutations in horses with white coat colour phenotypes. Anim. Genet. 42(3), 337–339. Haase, B., S. A. Brooks, A. Schlumbaum, P. J. Azor, E. Bailey, F. Alaeddine, M. Mevissen, D. Burger, P. A. Poncet, S. Rieder and T. Leeb. 2007. Allelic heterogeneity at the equine KIT locus in dominant white (W) horses." PLoS Genet 3(11), e195. Haque, B., Guirguis, G., Curtis, M., Mohsin, H., Walker, S., Morrow, M.M. and Costain, G. 2024. A comparative medical genomics approach may facilitate the interpretation of rare missense variation. J. Med. Genet. 61(8), 817–821. Hauswirth, R., Jude, R., Haase, B., Bellone, R.R., Archer, S., Holl, H., Brooks, S.A., Tozaki, T., Penedo, M.C.T., Rieder, S. and Leeb, T. 2013. Novel variants in the KIT and PAX3 genes in horses with white-spotted coat colour phenotypes. Anim. Genet. 44(6), 763–765. Nicholas, F. W., Tammen, I. and Sydney Informatics Hub. 2025. Online Mendelian Inheritance in Animals (OMIA). Pham, D.D.M., Guhan, S. and Tsao, H. 2020. KIT and melanoma: biological insights and clinical implications. Yonsei Med. J. 61(7), 562–571. Rexroad, C., Vallet, J., Matukumalli, L.K., Reecy, J., Bickhart, D., Blackburn, H., Boggess, M., Cheng, H., Clutter, A., Cockett, N., Ernst, C., Fulton, J.E., Liu, J., Lunney, J., Neibergs, H., Purcell, C., Smith, T.P.L., Sonstegard, T., Taylor, J., Telugu, B., Eenennaam, A.V., Tassell, C.P.V. and Wells, K. 2019. Genome to phenome: improving animal health, production, and well-being–a new USDA blueprint for animal genome research 2018–2027. Front. Genet. 10, 327. Spritz, R.A., Droetto, S. and Fukushima, Y. 1992. Deletion of the KIT and PDGFRA genes in a patient with piebaldism. Am. J. Med. Genet. 44(4), 492–495. Yang, Y., Shao, A. and Vihinen, M. 2022. PON-All: amino acid substitution tolerance predictor for all organisms. Front. Mol. Biosci. 9, 867572. Zhong, H., Zhang, J., Tan, C., Shi, J., Yang, J., Cai, G., Wu, Z. and Yang, H. 2022. Pig coat color manipulation by MC1R gene editing. Int. J. Mol. Sci. 23(18). Supplementary materialsTable S2. Selected prediction performance information for AlphaMissense, ESM-1b, and PolyPhen-2 (overall).
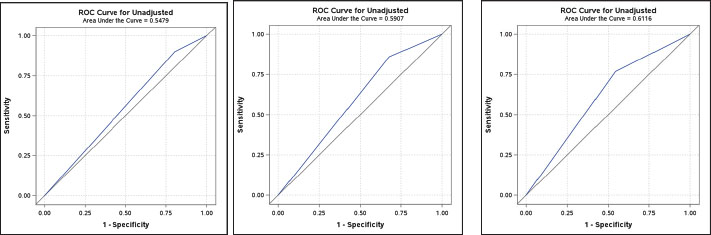
Fig. S1. Unadjusted receiver operating characteristic (ROC) curves for overall group for AlphaMissense (A), ESM-1b (B), and PolyPhen-2 (C).
Fig. S2. (A) Species effects on pathogenicity prediction of missense variants in domestic mammals (logistic regression, N=374, ESM-1b) and (B) species effects on pathogenicity prediction of missense variants in domestic mammals (logistic regression, N=368, PolyPhen-2). ESM-CLASS_NUM and PolyPhen-2_CLASS_NUM=predicted pathogenicity status.
Fig. S3. (A) Species-specific performance of PolyPhen-2 and (B) species-specific performance of ESM-1b: sensitivity, specificity, and auROC values for cattle, cats, dogs, horses, and sheep. | ||
| How to Cite this Article |
| Pubmed Style Hu H, Xie W. Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy. Open Vet. J.. 2025; 15(10): 5183-5191. doi:10.5455/OVJ.2025.v15.i10.35 Web Style Hu H, Xie W. Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy. https://www.openveterinaryjournal.com/?mno=268109 [Access: January 25, 2026]. doi:10.5455/OVJ.2025.v15.i10.35 AMA (American Medical Association) Style Hu H, Xie W. Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy. Open Vet. J.. 2025; 15(10): 5183-5191. doi:10.5455/OVJ.2025.v15.i10.35 Vancouver/ICMJE Style Hu H, Xie W. Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy. Open Vet. J.. (2025), [cited January 25, 2026]; 15(10): 5183-5191. doi:10.5455/OVJ.2025.v15.i10.35 Harvard Style Hu, H. & Xie, . W. (2025) Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy. Open Vet. J., 15 (10), 5183-5191. doi:10.5455/OVJ.2025.v15.i10.35 Turabian Style Hu, Hanwen, and Wei Xie. 2025. Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy. Open Veterinary Journal, 15 (10), 5183-5191. doi:10.5455/OVJ.2025.v15.i10.35 Chicago Style Hu, Hanwen, and Wei Xie. "Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy." Open Veterinary Journal 15 (2025), 5183-5191. doi:10.5455/OVJ.2025.v15.i10.35 MLA (The Modern Language Association) Style Hu, Hanwen, and Wei Xie. "Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy." Open Veterinary Journal 15.10 (2025), 5183-5191. Print. doi:10.5455/OVJ.2025.v15.i10.35 APA (American Psychological Association) Style Hu, H. & Xie, . W. (2025) Human genomic tools to assess missense variant pathogenicity in domestic mammals using a mock-variant strategy. Open Veterinary Journal, 15 (10), 5183-5191. doi:10.5455/OVJ.2025.v15.i10.35 |

