




| Short Communication | ||
Open Vet. J.. 2020; 10(1): 53-58 doi: 10.4314/ovj.v10i1.9 Open Veterinary Journal, (2020), Vol. 10(1): 53–58 Short Communication DOI: http://dx.doi.org/10.4314/ovj.v10i1.9 Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCRHannes Beims1,2*, Martina Janke1, Werner von der Ohe1 and Michael Steinert21Lower Saxony State Office for Consumer Protection and Food Safety, Institute of Apiculture, Celle 29221, Germany 2Institut für Mikrobiologie, Technische Universität Braunschweig, Braunschweig 38106, Germany *Corresponding Author: Hannes Beims. Lower Saxony State Office for Consumer Protection and Food Safety, Institute of Apiculture, Celle 29221, Germany. Email: Hannes.Beims [at] LAVES.Niedersachsen.de Submitted: 21/05/2019 Accepted: 14/02/2020 Published: 13/03/2020 © 2020 Open Veterinary Journal
AbstractBackground: American Foulbrood (AFB) is a devastating disease of honey bee (Apis mellifera) larvae caused by the spore-forming, Gram-positive bacterium Paenibacillus larvae. In most countries, the law requires mandatory reporting of AFB to the veterinary authority. Aim and Methods: To speed up detection and genotyping of P. larvae spores, we compared different culturing protocols on Columbia sheep blood agar and developed a new multiplex quantitative polymerase chain reaction to distinguish between the two relevant P. larvae genotypes enterobacterial repetitive intergenic consensus (ERIC) I and ERIC II. Results and Conclusion: As confirmed by P. larvae reference strains and field isolates, the new identification and genotyping protocol halves the time of current workflows, lessens labor-intension, allows a higher throughput of samples for monitoring, and permits a faster intervention to prevent the spread of AFB. Keywords: American Foulbrood, ERIC genotyping, Multiplex quantitative PCR, Paenibacillus larvae, Rapid detection. IntroductionPaenibacillus larvae, a Gram-positive, rod-shaped, flagellated and spore-forming bacterium, is the etiological agent of American Foulbrood (AFB), a deadly brood disease of honey bees (Apis mellifera) (Genersch, 2010). The infection starts with the oral uptake of P. larvae endospores by bee larvae below 36 hours of age (Yue et al., 2008). The transmission of the extremely resilient and long-lived spores within or between bee colonies occurs by contaminated adult bees and spore-containing larval food. However, bee keepers contribute to the spread of AFB by feeding contaminated honey, moving diseased hives, or using contaminated equipment (von der Ohe et al., 1996, 1997a, 1997b; Genersch, 2010). The P. larvae spores remain infectious for several decades, are resistant to heat and antibiotics, and about 10 are sufficient to infect and finally kill a larva (Woodrow and Holst, 1942; Genersch, 2010). After germination of spores, vegetative cells of P. larvae massively proliferate in the larval midgut and later invade the haemocoel. The bee larvae subsequently disintegrate into a ropy mass which dries down to a spore-containing, highly contagious scale. With the progress of the disease fewer bees reach adulthood and the population of the hive collapses (Yue et al., 2008; Genersch, 2010; Fünfhaus et al., 2013). Until now five genotypes, P. larvae enterobacterial repetitive intergenic consensus (ERIC) I–V, have been identified based on ERIC sequences (Genersch and Otten, 2003; Genersch et al., 2006; Beims et al., 2020). While P. larvae I and II are frequently isolated from AFB-infected colonies, the genotypes ERIC III and IV have not been isolated in the recent years (Genersch, 2010; Ebeling et al., 2016), whereas genotype ERIC V was isolated in 2013 from a honey sample, without having information about the effected colonies (Beims et al., 2020). ERIC I and II isolates differ in endospore resistance, rate of sporulation, and the strategy and time they need to kill their hosts (Genersch and Otten, 2003; Genersch et al., 2005, 2006; Poppinga et al., 2012; Fünfhaus et al., 2013; Beims et al., 2015; Morrissey et al., 2015). Therefore, it is crucial to discriminate the slow killing phenotype of ERIC I (12 days) and the fast killing phenotype of ERIC II (7 days) (Genersch, 2010). Moreover, genotyping is necessary to refine AFB epidemiology and outbreak management (Morrissey et al., 2015). However, methods for genotyping like MLVA still exist (Descamps et al., 2016), but the main routine diagnostic is based on P. larvae detection without any epidemiological information. Our method allows to distinguish between the slow (ERIC I) and the fast killing (ERIC II-V) genotypes, which increases the diagnostic information. The high contagiousness and the severity of AFB are the reasons why this most devastating bacterial infection of honeybees is subjected to registration in many countries. Since the current treatment options for bee colonies suffering from AFB are very limited, it is very important to timely identify outbreaks and to confirm the source and routes of disease transmission (von der Ohe et al., 1997a, 1997b; Morrissey et al., 2015). Traditionally, the glue-like larval remains that can be drawn out as a thread with a matchstick represent a first-line warning practice of beekeepers (De Graaf et al., 2006). Further clinical symptoms of AFB are diverse and depend on the genotype, the stage of disease, and the strength of the bee colony. Visible signs may include a patchy brood pattern, concave and perforated cell cappings, and flat scales tightly attached to the lower brood cell wall. Moreover, severely infected colonies emit a rotten odor (De Graaf et al., 2006). However, obvious symptoms often need up to 3 years to develop and similar clinical signs can result from other bee diseases (von der Ohe et al., 1996, 1997a, 1997b). According to this pathogenic development, colonies can be separated into uninfected ones, which do not harbor spores of P. larvae, and infected ones. These infected colonies might not show any visual symptoms in the first stages of infection, whereas spores of P. larvae can be detected in different matrices of the hive, i.e., food and comb material. Not till then, when P. larvae spread among the colony and a critical number of spores is reached, typical symptoms of AFB can be detected. In the past decades, considerable progress has been made in the taxonomic reclassification of P. larvae and in the diagnosis of AFB (Ash et al., 1991; Govan et al., 1999; Genersch and Otten, 2003; Genersch et al., 2006; Morrissey et al., 2015; Ruiz-Villalba et al., 2017). Moreover, several sensitive and selective culture media and molecular techniques have been developed (De Graaf et al., 2006; Amtliche Methodensammlung, 2017; Rossi et al., 2018). Since cultural methods need confirmation by isolate identification, numbers of 16S rRNA-based polymerase chain reaction (PCR) protocols were established (Govan et al., 1999; Dobbelaere et al., 2001; OIE, 2008). However, these molecularbiological methods can only be used for the detection of vegetative P. larvae from larval remains or bacterial cultures. To specify these PCR-based protcols, we developed a multiplex quantitative PCR (mqPCR). This protocol allows reliable species identification by targeting 16S rRNA and ItuC gene sequences (Djukic et al., 2014; Sood et al., 2014). The ERIC genotyping is achieved by targeting gene sequences encoding for Plx1, an AB toxin which is present in ERIC I but not in ERIC II (Fünfhaus et al., 2013). Material and MethodsBacterial strains and culture conditionsAll of the bacterial strains used in this study are listed in Table 1. Bacterial reference strains were P. larvae DSM 7030 (ERIC I), DSM 25430 (ERIC II), DSM 17237 (ERIC II; RSK 16 standard) and P. alvei (DSM 29). Field isolates of P. larvae genotype ERIC I and II were originally derived from honey samples by the Lower Saxony State Office for Consumer Protection and Fodd Sefety, Institute of Apiculture, Celle, Germany (Beims et al., 2015). If not stated otherwise, all strains were cultivated on house-made Columbia sheep blood agar (CSA-LAVES) at 37°C (https://www.dsmz.de/). Alternatively, purchased CSA agar plates from Oxoid or Becton Dickinson (BD) were used. Table 1. Resulting amplifications by verifying and genotyping P. larvae strains by multiplexed qPCR with different targets by their “final call (ΔRn)”. –, no amplification of the taget; +, amplification of the target.
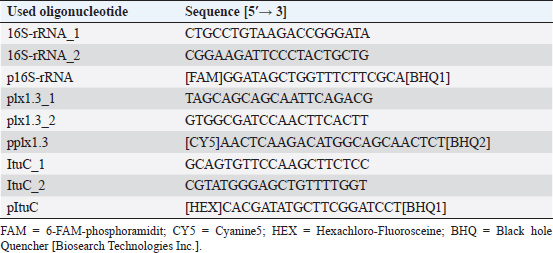
Germination of P. larvae spores on different preparations of CSAGermination assays were performed with spore suspensions from P. larvae DSM 17237 (RSK16 standard; LAVES IB CE). Aliquots of 200 µl RSK 16 standard were spread on CSA-LAVES (homemade) and commercially available CSA-Oxoid (Oxoid) and CSA-BD plates BD plates. Germination rates of spores were quantified and compared after 3 and 6 days. The mean and its standard error were determined. Significance was tested by Student’s t-test. Multiplex quantitative PCR (mqPCR) for P. larvaeMaterial from P. larvae colonies was homogenized in 50 µl sterile, deionized water, and incubated at 95°C for 15 minutes and 250 rpm. To sediment the bacterial debris, the mixture was centrifuged for 5 minutes at 5,000 × g. The supernatant was then used as template for mqPCR. The sequences of primers and probes targeting P. larvae are shown in Table 2. Oligonucleotides were designed based on the 16S rRNA sequence of P. larvae and the ItuC and Plx1 encoding sequences (http://bioinfo.ut.ee/primer3-0.4./) using the genome data of P. larvae DSM 7030 and DSM 25430 (Fünfhaus et al., 2013; Djukic et al., 2014; Sood et al., 2014; Hertlein et al., 2016; Rossi et al., 2018). The mqPCR reactions were performed in a total volume of 20 µl containing 10 µl 2× Luna Universal Probe qPCR Mix (NEB, Germany), 0.4 µM of each primer (eurofins genomics, Germany), 0.2 µM of each probe (eurofins genomics, Germany), 2.0 µl template and nuclease-free water to reach the reaction volume. The amplification was performed in an ARIA MX qPCR thermocycler (Agilent, USA), according to the manufacturer instruction (initial denaturation 95.0°C; 40 cycles: 95.0°C for 15 seconds and 60.0°C for 30 seconds, including detection of amplification at the end of the 60.0°C step). A no template control (NTC) was used, as well as P. larvae reference strains for genotype ERIC I and ERIC II (positive controls), and P. alvei as negative control. Amplification was analyzed using the Aria Agilent 1.3 software (Agilent, USA). Targets were interpreted as positive (“detected”) when Finall Call (ΔRn) was marked as “+” by the software, with a quantification cycle (Cq) (ΔRn) ≤ 30 (Ruiz-Villalba et al., 2017). Table 2. Primer and Probes used for detection and genotyping of P. larvae by multiplexed qPCR.
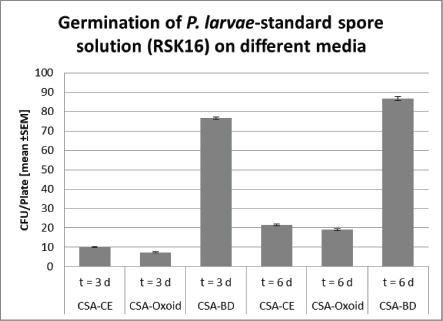
Ethical approvalAll experiments were performed according to actual ethical guidelines. Results and DiscussionTraditionally, monitoring of AFB is based on the observation of clinical symptoms within the apiary and microbial cultivation of P. larvae from honey, food, and brood samples (De Graaf et al., 2006). All cultivation protocols for the detection of sub-clinical levels of the pathogen include a heat treatment of the samples to reduce contamination and to stimulate the germination of P. larvae spores (Forsgren et al., 2008). Moreover, Nalidixid Acid can be used to avoid the growth of Gram-negative bacteria (Amtliche Methodensammlung, 2017). In our process, we reduced Gram-negative contaminants by a sample-processing step at 95°C. However, differences in heat resistance and germination rate of spores, as well as variation in proliferation among different P. larvae genotypes and strains may lead to a biased diagnose and disease monitoring (Forsgren et al., 2008). Not surprisingly, a high variability of bacterial plate counts from infected colonies has been reported (von der Ohe et al., 1997a, 1997b). Since selection of the growth media may also influence quantification of the pathogen (Nordström and Fries, 1995), we compared the germination of P. larvae spores on CSA, because of its ready-to-use disposability, instead of other media like MYPGP. Three different CSA without Nalidixid Acid were tested for its sensitivity against P. larvae. Therefore, we used freshly prepared CSA-LAVES and purchased CSA-Oxoid and CSA-BD plates, respectively (Fig. 1). The average germination rate of P. larvae DSM 17237 spores (RSK 16 standard) revealed moderate differences on CSA-LAVES (n=123) and CSA-Oxoid (n=120) after 3 and 6 days, respectively. After 3 days of incubation germination on CSA-LAVES (10.1 ±0.33 cfu/plate) and CSA-Oxoid (7.39 ± 0.27 cfu/plate) plates differed by factor 0.7 (p=1.44 × 10–9). In contrast, the bacterial plate counts on CSA-BD (n=120) were much higher at both time points. Germination on CSA-LAVES and CSA-BD (76.32 ± 0.68 cfu/plate) plates differed by factor 7.6 (p < 2.2 × 10–16) (Fig. 1). After 6 days of incubation CSA-LAVES (21.49 ±0.44 cfu/plate) and CSA-Oxid (19.19 ± 0.41 cfu/plate) differed by factor 0.9 (p=1.71 × 10–4), whereas CSA-LAVES and CSA-BD (86.61 ± 1.05 cfu/plate) differed by factor 4 (p < 2.2 × 10–16) (Fig. 1). The moderate differences between CSA-LAVES and CSA-Oxoid may be explained with the fresh sheepblood used for CSA-LAVES or the age of the purchased agar plates. However, the efficient germination and growth rate on CSA-BD suggests that additional ingredients of this complex medium are responsible for the marked difference. This is supported by our observation that cfu values of P. larvae on CSA-LAVES and CSA-Oxoid media are increasing over a time period of 6 days, while significantly higher cfu values on CSA-BD are already present after 3 days of cultivation. Thus, the faster and higher germination and growth rate after only 3 days suggests CSA-BD for a time-optimized protocol. To timely verify culture-based identification of P. larvae strains and to differentiate between the epidemiologically prevalent P. larvae genotypes ERIC I and II, we developed and optimized a multiplex qPCR. With this protocol P. larvae reference strains (DSM 7030 ERIC I, DSM 25430 ERIC II) and 40 field isolates could be confirmed based on the amplification of the 16S rRNA target sequence (Cq-value ≤ 30) (Table 1), while no amplicons were detected for P. alvei (no Cq) and the NTC (Cq-value > 30). Similarly, PCR products of the ItuC target gene were detected for all P. larvae reference strains and field isolates (Cq-value ≤ 30). The NTC showed no Cq and P. alvei resulted in Cq-values greater or equal 35. Since P. larvae genotypes ERIC I and II differ in their virulence gene repertoir (Fünfhaus et al., 2013; Djukic et al., 2014; Sood et al., 2014; Ebeling et al., 2016), we utilized the ERIC I specific AB toxin Plx1 gene to discriminate between them in our mqPCR approach. As shown in Table 1, target sequence plx1.3 was only detected in P. larvae DSM 7030 (Cq ≤ 30) and the field isolates of genotype ERIC I. Paenibacillus larvae DSM 25430 and all field isolate of genotype ERIC II were negative for plx1.3, whereas the Cq-values of the NTC and P. alvei were greater or equal 35. Thus, together with the results for the 16S rRNA and ItuC target sequences P. alvei can clearly be excluded and ERIC genotypes I and II of P. larvae be distinguished.
Fig. 1. Germination of P. larvae-stanadard spore solution (RSK16) on different Columbia Sheepblood-Agar. Significance was calculated by Student’s t-test (***, p < 0.001).
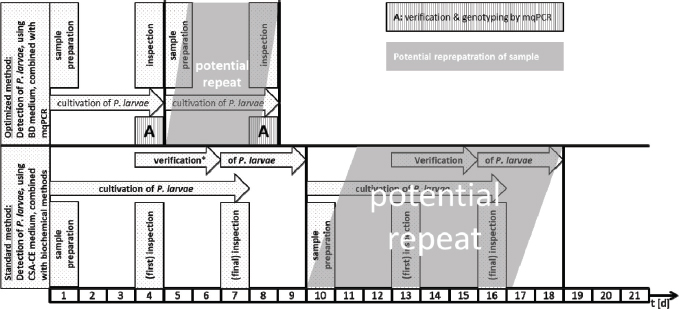
Fig. 2. Schematic comparison of the current standard culture-based method to detect P. larvae in honey and food samples and the newly developed protocol which combines optimized culture and mqPCR. ConclusionBy combining a culture-based approach on CSA-BD with multiplex qPCR, we were able to detect and genotype P. larvae reference strains and field isolates within 4 days (Fig. 2). This is half of the time needed with the actual official standard protocol (Amtliche Methodensammlung, 2017). Since time consuming methods, such as purification of bacterial DNA, gel electrophoresis, and interpretation of fragment patterns are not necessary, the newly developed multiplex qPCR allows a higher throughput of samples, may reduce costs for monitoring surveys, and permit faster interventions to prevent the spread of AFB. AcknowledgmentsThe authors would like to thank Katlen Schütze, Florian Hinz and Friewi Lienau for excellent technical assistance. Author’s contributionsHannes Beims designed the experiments for mqPCR and was involved in data analysis of germination assays. Martina Janke and Werner von der Ohe provided the germination assays and verified analytical methods. Michael Steinert analyzed the derived data. All authors discussed the results and contributed to the final manuscript. ReferencesAmtliche Methodensammlung. 2017. Amerikanische Faulbrut (Erreger: Paenibacillus larvae). Friedrich-Loeffler-Institut (FLI), Stand 09.06.2017, 7. Ash, C., Farrow, J.A.E., Wellbanks, S. and Collins, M.D. 1991. Phylogenetic heterogeneity of the genus Bacillus revealed by comparative analysis of small subunitribosomal RNA sequences. Lett. Appl. Microbiol. 13, 202–206. Beims, H., Bunk, B., Erler, S., Mohr, K.I., Spröer, C., Pradella, S., Günther, G., Rohde, M., von der Ohe, W. and Steinert, M. 2020. Discovery of Paenibacillus larvae ERIC V: Phenotypic and genomic comparison to genotypes ERIC I-IV reveal different inventories of virulence factors which correlate with epidemiological prevalences of American Foulbrood. Int. J. Med. Microbiol. 310(2), 151394. https://doi.org/10.1016/j.ijmm.2020.151394. Beims, H., Wittmann, J., Bunk, B., Spröer, C., Rhode, C., Günther, G., Rohde, M., von der Ohe, W. and Steinert, M. 2015. Paenibacillus larvae-directed bacteriophage HB10c2 and its application in American Foulbrood-affected honey bee larvae. Appl. Environ. Microbiol. 81(16), 5411–5419. De Graaf, D.C., Alippi, A.M., Brown, M., Evans, J.D., Feldlaufer, M., Gregorc, A., Hornitzky, M., Pernal, S.F., Schuch, D.M.T., Titera, D., Tomkins, V. and Ritter, W. 2006. Diagnosis of American foulbrood in honey bees: a synthesis and proposed analytical protocols. Lett. Appl. Microbiol. 43, 583–590. Descamps, T., De Smet, L., Stragier, P., De Vos, P. and de Graaf, D.C. 2016. Multiple locus variable number of tandem repeat analysis: a molecular genotyping tool for Paenibacillus larvae. Microb. Biotechnol. 9(6), 772–781. Djukic, M., Brzuszkiewicz, E., Fünfhaus, A., Voss, J., Gollnow, K., Poppinga, L., Liesegang, H., Garcia-Gonzalez, E., Genersch, E. and Daniel, R. 2014. How to kill the honey bee larva: Genomic potential and virulence mechanisms of Paenibacillus larvae. PLoS One 9, e90914. Dobbelaere, W., de Graaf, D.C., Peeters, J.E. and Jacobs, F.J. 2001. Development of a fast and reliable diagnostic method for American foulbrood disease (Paenibacillus larvae subsp. Larvae) using 16S rRNA gene based PCR. Apidologie 32, 363–370. Ebeling, J., Knispel, H., Hertlein, G. and Fünfhaus, A. 2016. Biology of Paenibacillus larvae, a deadly pathogen of honey bee larvae. Appl. Microbiol. Biotechnol. 100, 7387–7395. Forsgren, E., Stevanovic, J. and Fries, I. 2008. Variability in germination and in temperature and storage resistance among genotypes. Vet. Microbiol. 129(3–4), 342. Fünfhaus, A., Poppinga, L. and Genersch, E. 2013. Identification and characterization of two novel toxins expressed by the lethal honey bee pathogen Paenibacillus larvae, the causative agent of American foulbrood. Environ. Microbiol. 15(11), 2951–2965. Genersch, E. 2010. American Foulbrood in honeybees and its causative agent, Paenibacillus larvae. J. Invertebr. Pathol. 103, 10–19. Genersch, E. and Otten, C. 2003. The use of repetitive element PCR fingerprinting (rep-PCR) for genetic subtyping of German field isolates of Paenibacillus larvae subsp. larvae. Apidologie 34, 195–206. Genersch, E., Ashiralieva, A. and Fries, I. 2005. Strain- and genotype-specific differences in virulence of Paenibacillus larvae subsp. larvae, a bacterial pathogen causing American foulbrood disease in honeybees. Appl. Environ. Microbiol. 71, 7551–7555. Genersch, E., Forsgren, E., Pentikäinen, J., Ashiralieva, A., Rauch, S., Kilwinski, J. and Fries, I. 2006. Reclassification of Paenibacillus larvae subsp. pulvifaciens and Paenibacillus larvae subsp. larvae as Paenibacillus larvae without subspecies differentiation. Int. J. Syst. Evol. Microbiol. 56, 501–511. Govan, V.A., Allsopp, M.H. and Davison, S. 1999. A PCR detection method for rapid identification of Paenibacillus larvae. Appl. Environ. Microbiol. 65(5), 2243–2245. Hertlein, G., Seiffert, M., Gensel, S., Garcia-Gonzales, E., Ebeling, J., Skobalj, R., Kuthning, A., Süssmuth, R.D. and Genersch, E. 2016. Biological role of paenilarvins, Iturin-like lipopeptide secondary metabolites produced by the honey bee pathogen Paenibacillus larvae. PLoS One. 11(10): e0164656. doi:10.1371/journal.pone.0164656. Morrissey, B.J., Helgason, T., Poppinga, L., Fünfhaus, A., Genersch, E. and Budge, E. 2015. Biogeography of Paenibacillus larvae, the causative agent of American foulbrood, using a new multilocus sequence typing scheme. Environ. Microbiol. 17(4), 1414–1424. Nordström, S. and Fries, I. 1995. A comparison of media and cultural conditions for identification of Bacillus larvae in honey. J. Apicult. Res. 34, 97–103. OIE. 2008. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 6th ed. Charpter 2.9.2 American Foulbrood. ISBN 978-92-9044-718-4. Poppinga, L., Janesch, B., Fünfhaus, A., Sekot, G., Garcia-Gonzalez, E., Hertlein, G., Hedtke, K., Schäffer, C. and Genersch, E. 2012. Identification and functional analysis of the S-Layer protein SplA of Paenibacillus larvae, the causative agent of American foulbrood of honey bees. PLoS Path. 8(5), e1002716. Rossi, F., Amadoro, C., Ruberto, A. and Ricchiuti, L. 2018. Evaluation of quiantitative PCR (qPCR) Paenibacillus larvae targeted assays and definition of optimal conditions for its detection/quantification in honey and hive debris. Insects 9, 165. Ruiz-Villalba, A., van Pelt-Verkuil, E., Gunst, Q.D., Ruijter, J.M. and van den Hoff, M.J.B. 2017. Amplification of nonspecific products in quantitative polymerase chain reactions (qPCR). Biomol. Detect. Quantif. 14, 7–18. Sood, S., Steinmetz, H., Beims, H., Mohr, K.I., Stadler, M., Djukic, M., von der Ohe, W., Steinert, M., Daniel R. and Müller, R. 2014. Paenilarvins, iturin family lipopeptides from the honey bee pathogen Paenibacillus larvae. Chembiochem 15(13), 1947–1955. Von der Ohe, W., Schütze, K. and Lienau, F.W. 1996. Arealuntersuchungen auf Bacillus-larvae-Sporen im Honig las Prophylaktikum. Apidologie 27(4), 277–279. Von der Ohe, W., Schütze, K. and Lienau, F.W. 1997a. Möglichkeiten der Vorbeugung der Amerikanischen Faulbrut mit Hilfe der Untersuchung von Futterproben. Die Biene 133(3), 8–10. Von der Ohe, W., Schütze, K. and Lienau, F.W. 1997b. Prophylaxe der Amerikanischen Faulbrut mit Hilfe der Untersuchung von Futterkranzproben auf Paenibacillus-larvae-larvae-Sporen. Deutsches Bienenjournal 5(4), 138–141. Woodrow, A.W. and Holst, E.C. 1942. The mechanism of colony resistance to American foulbrood. J. Econom. Entomol. 35, 327–330. Yue, D., Nordhoff, M., Wieler, L.H. and Genersch, E. 2008. Fluorescence in situ hybridization (FISH) analysis of the interactions between honeybee larvae and Paenibacillus larvae, the causative agent of American Foulbrood of honeybees (Apis mellifera). Environ. Microbiol. 10(6), 1612–1620. | ||

| How to Cite this Article |
| Pubmed Style Beims H, Janke M, Ohe WVD, Steinert M. Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. Open Vet. J.. 2020; 10(1): 53-58. doi:10.4314/ovj.v10i1.9 Web Style Beims H, Janke M, Ohe WVD, Steinert M. Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. https://www.openveterinaryjournal.com/?mno=49844 [Access: June 26, 2026]. doi:10.4314/ovj.v10i1.9 AMA (American Medical Association) Style Beims H, Janke M, Ohe WVD, Steinert M. Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. Open Vet. J.. 2020; 10(1): 53-58. doi:10.4314/ovj.v10i1.9 Vancouver/ICMJE Style Beims H, Janke M, Ohe WVD, Steinert M. Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. Open Vet. J.. (2020), [cited June 26, 2026]; 10(1): 53-58. doi:10.4314/ovj.v10i1.9 Harvard Style Beims, H., Janke, . M., Ohe, . W. V. D. & Steinert, . M. (2020) Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. Open Vet. J., 10 (1), 53-58. doi:10.4314/ovj.v10i1.9 Turabian Style Beims, Hannes, Martina Janke, Werner Von Der Ohe, and Michael Steinert. 2020. Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. Open Veterinary Journal, 10 (1), 53-58. doi:10.4314/ovj.v10i1.9 Chicago Style Beims, Hannes, Martina Janke, Werner Von Der Ohe, and Michael Steinert. "Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR." Open Veterinary Journal 10 (2020), 53-58. doi:10.4314/ovj.v10i1.9 MLA (The Modern Language Association) Style Beims, Hannes, Martina Janke, Werner Von Der Ohe, and Michael Steinert. "Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR." Open Veterinary Journal 10.1 (2020), 53-58. Print. doi:10.4314/ovj.v10i1.9 APA (American Psychological Association) Style Beims, H., Janke, . M., Ohe, . W. V. D. & Steinert, . M. (2020) Rapid identification and genotyping of the honeybee pathogen Paenibacillus larvae by combining culturing and multiplex quantitative PCR. Open Veterinary Journal, 10 (1), 53-58. doi:10.4314/ovj.v10i1.9 |

