




| Research Article | ||
Open Vet. J.. 2024; 14(12): 3269-3288 Open Veterinary Journal, (2024), Vol. 14(12): 3269-3288 Research Article First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genesMohammed A. Al-Bukhalifa and Hassan M. Al-Tameemi*Microbiology Department, College of Veterinary Medicine, Basrah University, Basrah, Iraq *Corresponding Author: Hassan M. Al-Tameemi. Microbiology Department, College of Veterinary Medicine, Basrah University, Basrah, Iraq. Email: hisan.jasim [at] uobasrah.edu.iq Submitted: 17/08/2024 Accepted: 12/11/2024 Published: 31/12/2024 © 2024 Open Veterinary Journal
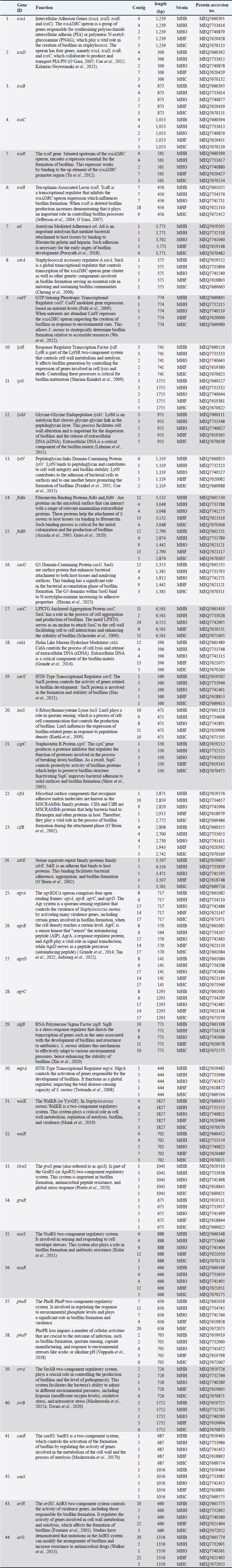
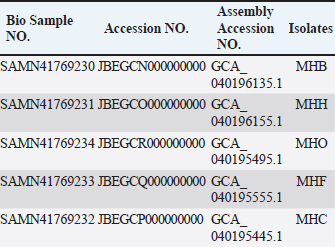
AbstractBackground: Staphylococcus aureus is a significant zoonotic pathogen capable of causing infections in both humans and animals. The bacterium’s capacity to develop biofilms and resistance to many different antibiotics has raised significant concerns for public health. Furthermore, studies have demonstrated that horizontal gene transfer enables the transfer of deleterious features between strains found in humans and animals, consequently rendering treatment and control efforts more challenging. Aim: This study aimed to investigate the relationships between human and animal isolates and biofilm-associated genes in local S. aureus strains using whole genome sequencing technique. Methods: We examined 111 suspected cases of S. aureus infection in humans and in animals and screened all S. aureus -positive isolates (11 isolates) for biofilm formation and antimicrobial profiles. Additionally, we sequenced and studied five S. aureus genomes isolated from humans, cows, sheep, cats, and dogs for significant biofilm-related genes and predicted their loci following annotation and deposition in the NCBI database. Results: The study showed that the isolates have genome sizes between 2.7 and 2.8 megabases, a GC content of 32.8%–33.1%, and a coding sequence count between 2,718 and 2,838. The cow isolate (MHB) and cat isolate (MHF) exhibited substantial genomic similarities with human isolates of S. aureus (N315) and the type strain of S. aureus (DSM 20231). The genomes of the human isolate (MHH) and the dog isolate (MHC) were comparable to S. aureus (N315). The sheep isolate (MHO) showed lesser genomic similarity and was closely related to S. aureus subsp. anaerobius. The genomes were submitted to the NCBI database with the following accession numbers: MHB (GCA_040196135.1), MHH (GCA_040196155.1), MHO (GCA_040195495.1), MHF (GCA_040195555.1), and MHC (GCA_040195445.1). The isolates were categorized by PubMLST typing into MHC (ST-1156), MHB (ST-6), MHF (ST-6), and MHO (a unique ST). We identified the accession numbers, locations, and lengths of biofilm-associated genes and regulators within the studied genomes. Conclusion: The study is the first to conduct complete genome sequencing of Staphylococcus aureus in Iraq, allowing analysis of biofilm-associated genes in local isolates. It provides the first large-scale genomic investigation of genetic relationships among animal and human isolates in Iraq. Keywords: Biofilm formation, Staphylococcus aureus, Whole genome sequencing, Zoonotic diseases. IntroductionStaphylococcus aureus is a significant pathogen known for causing a wide range of infections, from minor skin conditions to severe diseases such as pneumonia, endocarditis, and sepsis. Studies demonstrates that horizontal gene transfer and mutations enhance the virulence characteristics of human and animal strains, consequently complicating treatment and control measures (Narongpun et al., 2023). Staphylococcus aureus ability to develop resistance to antibiotics (especially methicillin resistant S. aureus (MRSA) and biofilms formation makes it particularly challenging threat (Archer, 1998; Becker, 2018; Ebani, 2020). Biofilms are organized communities of bacteria which are encased in a self-produced polymeric matrix, that adhere to surfaces and provide protection against hostile environments, including antibiotic treatment (Idrees et al., 2021). The biofilm-related genes in S. aureus play a central role in its ability to cause persistent in both hospital and community settings, contributing to chronic infections and increased resistance to conventional therapies (Arciola et al., 2012; Hong and Roh, 2018; Thiran et al., 2018). In order to counter diverse immune responses inside the host, pathogens need a wide range of sophisticated mechanisms that are associated with fast and efficient regulatory capacities (Thamer and Shareef, 2022). As a result, the regulatory network of S. aureus is quite complex and consists of many regulators that are instrumental in determining the best adaptive responses. Of these important regulators are the two-component systems (TCS) regulators. TCS are essential for bacterial survival and adaptation in various conditions. These TCSs are composed of a sensor kinase and a response regulator that can sense and react to different kinds of environmental signals, such as biofilm formation systems, antibiotic resistance, and virulence (Beier and Gross, 2006). Humans have been historically recognized as the primary transmission hub for S. aureus to other species. However, a variety of host jump events have facilitated the emergence of endemic livestock strains, and subsequent host switches back into humans have led to the expansion of global epidemic S. aureus clones. Genetic modifications (genetic drift) in the genomes of the adapted S. aureus isolates facilitated these host jumps (Rodrigues et al., 2022). Genetic drift, a mechanism of neutral diversification or adaptive evolution, may influence the host adaptability of S. aureus by favoring advantageous mutations and reducing detrimental ones in the new host. These genomic modifications require not only the ability to evade the immunity of the new host but also to be able to transmit the new clone infection between the new host group members (Rodrigues et al., 2022). For instance, single nucleotide polymorphisms and insertion-deletion mutations (indels) might broaden the host range of a S. aureus isolate, while the incorporation of mobile genetic components may introduce new genes into its genome, facilitating infection and survival in other hosts (Viana et al., 2015; Bacigalupe et al., 2019). These genomic modifications could occur in key host adaptation genes such as biofilm genes or antimicrobial genes (Aboud and Khudaier, 2018; Shareef et al., 2023). The immune system’s selective pressure and antimicrobial treatment may facilitate these genetic modification events during the infection course. Nevertheless, these alterations could only increase virulence, and they would not inherently alter host adaptation or host tropism, which may require in vivo experimental validation (Paharik and Horswill, 2016; Howden et al., 2023). In S. aureus infection, biofilm formation is a key process in adaptation against the host’s defense mechanisms, allowing this bacterium to colonize and adapt. Therefore, genetic variations in the genes and regulators associated with biofilm formation may expand the virulence and even host tropism, enabling the infection of new hosts (Howden et al., 2023). This recurrent genetic drift necessitates continuous monitoring and analysis of pathogen genomes. It is therefore important to understand the pathogen genomes as well as coding sequences (CDs) within the community for developing effective therapeutic approaches (Kwong et al., 2015; O’Connor et al., 2018; Humphreys and Coleman, 2019). Furthermore, it would be advantageous to examine the biofilm-associated genes in various host species of S. aureus isolates to identify potential genetic variations that may influence interspecies infection or the transmission of illnesses among animals (Howden et al., 2023). The efficiency and cost-effectiveness of next-generation sequencing have significantly improved, resulting in a transformational effect on several genomics-related fields (Brlek et al., 2024; Bagger et al., 2024). Consequently, we have annotated the genomes of local isolates to track any alterations in the local S. aureus isolates in forthcoming genomic investigations. This study aimed to investigate the relationships between human and animal isolates and biofilm-associated genes in local S. aureus strains using whole genome sequencing technique. We sequenced the genomes of five S. aureus isolates from clinical cases in humans and animals. These isolates exhibited a robust capacity for biofilm formation. We employed bioinformatic approach to determine the phylogenetic relationship among these isolates and subsequently identified the biofilm-associated genes within their genomes. This study represents the first application of whole genome sequencing (WGS) on S. aureus isolates in Iraq, potentially offering significant insights for further research endeavors. Materials and MethodsSamplesWe obtained samples of suspected S. aureus infections (N=111) from affected animals at veterinary clinics and hospitals in the Basrah governorate. This included pus and mastitis in animals. We obtained five human isolates from the Basrah Directorate Health Central Laboratory (identified S. aureus from pneumonia and pus cases). We focused on cases in animals exhibiting symptoms indicative of S. aureus infection, including pus and mastitis. The cases included: cows (25 cases of mastitis), sheep (20 cases of mastitis and five cases of pus), dogs (13 cases of pus), cats (12 cases of pus), camels (15 cases of pus), and chickens (15 cases of pus). We processed and cultivated each sample using standard microbiological techniques for isolation and identification, including growth on Mannitol salt agar, CHROMagarTM S. aureus, and Gram staining, before submitting it to the Vitek2 identification system. Molecular identificationUsing the universal primers 27F 5 ′- AGAGTTTGATCCTGGCTCAG-3 ′ and 1492R 5′-TACGGTTACCTTGTTACGACT-3′) (Frank et al., 2008; Srinivasan et al., 2015), the 1500 base pair PCR product of the 16 s RNA gene was sequenced using the internal sequencing primers the 27F and 907R 5’-CCGTCAATTCMTTTRAGTTT-3’, the 785F 5’-GGATTAGATACCCTGGTA-3’ and 1492R, respectively. All the sequencing processes were carried out at Macrogen , Korea. Blast analysis throughout the NCBI database was conducted to confirm the identity of each isolate (Marchler-Bauer et al., 2015; Tatusova et al., 2016; Yang et al., 2020). Biofilm formationWe investigated the biofilm formation as previously described, with slight modifications (Mashruwala et al., 2017a; Mashruwala et al., 2017b). We diluted aerobic cultures that were cultivated overnight in brain heart broth (obtained from individual colonies) into fresh sterile brain heart broths until the final optical density reached 0.05 (A630). 200 µl of diluted cultures were added to Costar® 96-well cell culture plates with a flat bottom (Corning , USA). The plates were incubated without agitation at 37°C for 22 hours. We evaluated the optical density (A630) of the cultures prior to harvesting the biofilm. The plates were washed three times with distilled water and then heat fixed at a temperature of 60°C for 1 hour. Finally, the plates were left to cool to room temperature before staining the biofilm mass with a 0.1% solution of crystal violet. Plates were washed three times with deionized water after staining. The biofilms were subsequently de-stained using a 33% acetic acid solution. After mixing the plates, the absorbance of the resulting solution was measured at 570 nm using BioTek 800TS microplate reader. The absorbance data were normalized to a blank acetic acid solution, and then to the harvested culture’s optical densities. Staphylococcus aureus MRSA ATCC 43300 and S. aureus ATCC 29213 were used as controls. Whole genome sequencingAfter the initial screening, we selected five S. aureus isolates for whole genome sequencing based on the intensity of their biofilm phenotypes. The isolates were named as follows: the S. aureus MHB from a mastitis case in a cow, the S. aureus MHO from a mastitis case in an ewe, the S. aureus MHF from ear infections in a cat, the S. aureus MHC from skin infections in a dog, and the S. aureus MHH from an upper respiratory infection in a human. The genomic DNA (gDNA) from these isolates was extracted using DNA extraction kit (QIAamp Mini Kit HB-0329 from Qiagen company, USA, catalog number 51304-51306) according to the manufacturer instruction. Prior to sequencing, the quality of the gDNA samples was assessed using nanodrop and gel electrophoresis. All the sequencing processes were carried out at Macrogen, Korea including the whole genome sequencing which was carried out using Illumina novaseq sequencer. The sequencing library was prepared by random fragmentation of the gDNA samples, followed by 5’ and 3’ adapter ligation. Adapter-ligated fragments are then PCR amplified with a PCR primer solution which anneals to the ends of each adapter (Keats et al., 2018; Maljkovic Berry et al., 2019). The library was loaded onto a flow cell at which fragments are seized on a lawn of surface-bound oligos corresponding to the library adapters. Each fragment was amplified into distinct clonal clusters via bridge amplification. the templates resulted from cluster generation were submitted for sequencing using Novaseq sequencer which generated raw reads in FASTQ files format. FastQC (0.11.7) was used to check the quality of raw sequence data(Andrews, 2010). The total number of bases, reads, GC (%), and Phred quality score Q20 (%), and Q30 (%) were calculated. Applying the Phred quality score at each cycle’s Q20 (%) and Q30 (%) helped assess the quality of the nucleobase identification provided by automated DNA sequencing, which in turn helped choose the quality of the data that was produced (Ewing and Green, 1998). Before the analysis, adapter sequences and low-quality bases were trimmed away from the reads using Trimmomatic program- version 0.38 (Bolger et al., 2014). In order to map the reads obtained from sequencing, S. aureus (NZ_AP014921.1) was used as a reference genome using BWA—Burrows-Wheeler Aligner (Li, 2013). After read mapping, Picard and SAMTools were utilized to remove duplicate reads and to determine variant information. Contigs were created by de novo assembly of the raw reads using the bioinformatics program SPAdes v.3.5. (Bankevich et al., 2012). Genome submissions to NCBI GenBankThe assembled genome sequences were deposited at GenBank Bio project PRJNA1121204. The annotation was added by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) (Tatusova et al., 2016). Multi locus sequence typing (MLST)We utilized the MLST server, software version 2.0.9 (2022-05-11) and database version 2023-06-19, provided by the Center for Genomic Epidemiology at https://www.genomicepidemiology.org. This server determines the sequence types (STs) of bacteria from the whole genomes sequence data (Larsen et al., 2012). Furthermore, the bacterial population genomics BIGSdb (PubMLST website) (Jolley and Maiden, 2010; Jolley et al., 2018) was used to confirm and index the sequence types of our isolates. Genome analysis and comparisonWe used PATRIC’s comprehensive genome analysis tool to examine the sequencing reads as previously described (Wattam et al., 2017). This helped to detect both conserved sequence features and produced approximate circular representationss of the genome using the circular genome viewer at the Bacterial and Viral Bioinformatics Resource Center (BV-BRC) website (Olson et al., 2023). Additionally, we further investigated the assembled genomes for detailed information on functional genes categorized within the subsystem groups using the RASTtk server (Brettin et al., 2015) and the SEED tool (Overbeek et al., 2014). For consistency, the minimal cutoff for similarity in all annotation and comparison methods was 95% identity. We used the SEED tool to compare and analyze the genomes of the animals to the human MHH strain, which served as the reference genome. Phylogenetic analysisWe submitted the genome sequences to the type (Strain) genome server (TYGS), a free bioinformatics platform at https://tygs.dsmz.de, for a whole genome taxonomy analysis. The List of Prokaryotic Names withstanding in Nomenclature (LPSN) database (available via https://lpsn.dsmz.de), offered information on nomenclature, synonymy, and relevant taxonomic references (Meier-Kolthoff et al., 2022). Results were provided by TYGS on 2024-07-15. Based on PATRIC’s comprehensive genome analysis phylogenic initial results, we included S. aureus subsp. aureus NCTC 8325 and S. aureus subsp. aureus N315 as controls in the TYG phylogenetic analysis, aiming to increase the resolution. Ethical approvalSampling from animals was conducted according to the ARRIVE guidelines and in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments. The human samples were collected from local hospitals’ laboratories under the supervision of the Basrah Directorate of Health and a written consent was signed by each patient. The research ethics committee of Basrah College of Veterinary Medicine granted this research the permission number 40/37/2024 on May 22, 2024. ResultsIsolation of S. aureusAfter identification, we evaluated 11 confirmed positive samples for S. aureus for biofilm production and antimicrobial properties, selecting five isolates for whole genome sequencing based on their biofilm-forming capacity and antibiotic resistance. We recognized these isolates as S. aureus, derived from human (MHH), cow (MHB), sheep (MHO), cat (MHF), and dog (MHC) origins. Genomes informationPhred scores of 20 (Q20) quantify the precision of the DNA sequencing technique. The sequencing quality was excellent ranging between Q20 (%) 98.1 to 98.3%. The genome sizes ranged from 2.7 to 2.8 megabase (Mb), the GC content percentage was 32.8-33.1 %, and the number of CDs ranged from 2,718 to 2,838 (Fig. 1, Table 1). The assembled genome sequences were deposited at GenBank Bio project PRJNA1121204. The annotation was added by the NCBI PGAP (Tatusova et al., 2016). In addition, the annotation process included an analysis of the subsystems unique to these genomes. The genomes were submitted to the NCBI database with the following accession numbers: MHB (GCA_040196135.1), MHH (GCA_040196155.1), MHO (GCA_040195495.1), MHF (GCA_040195555.1), and MHC (GCA_040195445.1). Figure 2 presents an overview of the S. aureus subsystem categories for MHH, MHB, MHC, MHF, and MHO strains. The bioinformatic analysis revealed no plasmids in any of the tested isolates. Multi locus sequence typingUsing the bacterial population genomics BIGSdb software and the PubMLST website (Jolley et al., 2018) we determined that the isolates belong to MHC ST-1156, MHB ST-6, MHF ST-6, whereas MHO is a new novel ST that is close to ST 522. Genome comparison and phylogenetic analysisThe RASTtk server and Seed Viewer sequence-based comparison tool revealed that while there were variations in the genes of the animals’ isolates, their genomes exhibited significant similarities (Fig. 3) and formed a cluster as species and subspecies in the phylogenetic tree based on the TYGS result for the local S. aureus whole-genome data set (Fig. 4). The genomes of cow isolate MHB and cat isolate MHF showed significant homology to the human isolates of S. aureus (N315) MRSA (Kuroda et al., 2001) and the type strain of S. aureus (DSM 20231) (Shiroma et al., 2015). The genomes of the human isolate MHH and the dog isolate MHC were remarkably comparable to those of S. aureus (N315). Nevertheless, the MHO strain exhibited a lesser degree of similarity in comparison to the human and other animals isolates and was close to S. aureus subsp. anaerobius (Elbir et al., 2013). The findings of our study support the presence of a strong phylogenetic connection between the human and animal isolates that were investigated. These findings provide further evidence that these isolates have a zoonotic origin. Upon comparing the genomes of animal isolates to the human strain MHH, we noticed dispersed groups of genes that either did not have any similarity or exhibited lower similarity when compared to the human isolate MHH (Fig. 3, marked with green, purple, blue, and orange arrows). Biofilm associated genesWe conducted a comprehensive review of the literature on S. aureus biofilm-associated genes and identified the sequences of 24 confirmed genes. Utilizing the NCBI BLAST analysis tool, we compared these sequences against the genomes of MHH, MHB, MHC, MHF, and MHO strains. Table 2 provides the accession numbers for the biofilm-associated genes in the studied genomes. Figure 5 shows a crystal violet assay for biofilm formation in S. aureus local clinical isolates MHB, MHC, MHH, MHF, and MHO. DiscussionWe selected five S. aureus isolates for whole genome sequencing based on the intensity of their biofilm phenotypes. The isolates were named as follows: S. aureus MHB from mastitis in cows, S. aureus MHO from mastitis in sheep, S. aureus MHF from ear infections in a cat, S. aureus MHC from skin infections in a dog, and S. aureus MHH from upper respiratory infections in a human. Prior to the start of this work, there were no genomic data sets available for Iraqi S. aureus isolates. The sole exceptions were S. xylosus (Al-Tameemi et al., 2023) and S. epidermidis (Talat et al., 2020). The purpose of this study was to sequence the whole genome of five local S. aureus isolates in order to identify and label genes involved in biofilm formation and to examine the potential zoonotic relationship between these organisms and animals. The findings will provide a strong basis for future research in this area. Therefore, our findings will expand the pool of genomes accessible from the Middle East, possibly explaining the variety of strains present in the region.
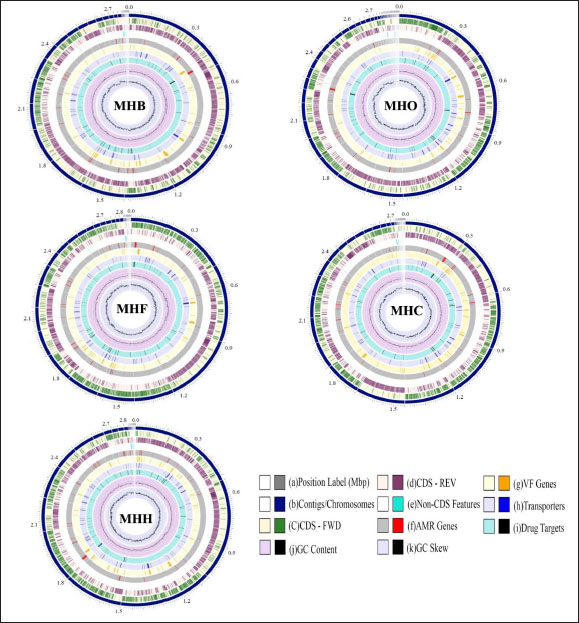
Fig. 1. A circular view of the genomes of S. aureus MHB, MHF, MHH, MHO, and MHC isolates generated by PATRIC (Wattam et al., 2017; Olson et al., 2023) shows the physical map of its important features. From outside in: (a) position in Mbp; (b) order of contigs (shown in navy); (c)/(d) distribution of forward and reverse CDs in plus and minus strands (shown in green and purple, respectively); (e) distribution of noncoding elements (shown in blue); (f) distribution of genes involved in antibiotic resistance (shown in red); (g) distribution of virulence factors (shown in orange); (h) distribution of genes encoding transmembrane proteins (shown in dark blue); (i) distribution of genes encoding drug targets (shown in black); (j) distribution of GC content along plus and minus strands; (k) GC skew (most inner two circles, respectively). Table 1. Genomes information for S. aureus isolates from human (MHH), cow (MHB), sheep (MHO), cat (MHF), and dog (MHC).
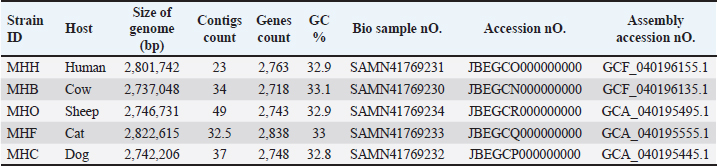
Fig. 2. The distribution of Staphylococcus aureus subsystem categories for MHH, MHB, MHC, MHF, and MHO strains. The genomes were annotation using the Rapid Annotation System Technology (RASTtk) tool. The pie chart displayed the number of each subsystem feature, while the SEED viewer showed the degree of subsystem coverage. The green bar in the subsystem coverage indicates the proportion of proteins that are part of the subsystems, whereas the blue bar reflects the proportion of proteins that are not part of the subsystems. The relationship between humans and animals has resulted in a significant rise in global incidences of zoonotic infections caused by bacteria that were previously known to infect only specific hosts (Bartels et al., 2021). Staphylococcus aureus is able to infect new hosts and cross species barriers, despite the fact that humans are considered its primary reservoir. Host-jumping events might occur between humans and animals and vice versa. Horizontal gene transfer of genetic elements that confer survival characteristics in new hosts has been associated with host-species transformations (Richardson et al., 2018). These transmissions between species have led to the rise of S. aureus lineages that can cause outbreaks and spread widely in both humans and animals. Understanding the population and genomic patterns of MRSA across various hosts is crucial for advancing a One Health strategy and securing optimal health for humans, domestic animals, wildlife, and the environment (Rodrigues et al., 2022). In S. aureus infection, biofilm formation is a key process in adaptation against the host’s defense mechanisms, allowing this bacterium to colonize and adapt (Mazaal et al., 2021). Therefore, genetic variations in the genes and regulators associated with biofilm formation may expand the virulence and even host tropism, enabling the infection of new hosts (Howden et al., 2023) . Consequently, we have annotated the genes in the genomes of local S. aureus isolates to track any alterations in the local S. aureus isolates in forthcoming genomic or epidemiological investigations. Due to the constant transfer of virulence genes through horizontal gene transfer among S. aureus populations, the flow of genomic information plays a crucial role in understanding and controlling this pathogenic bacterium. The prolonged instability and conflicts in Iraq have hindered the ability to carry out extensive research that can track alterations in the genetic composition of persistent pathogenic infections such as S. aureus. To the best of our knowledge, only a two gram-positive Staphylococcus whole genome sequencing studies have been conducted in Iraq, such as S. xylosus (Al-Tameemi et al., 2023) and S. epidermidis (Talat et al., 2020).
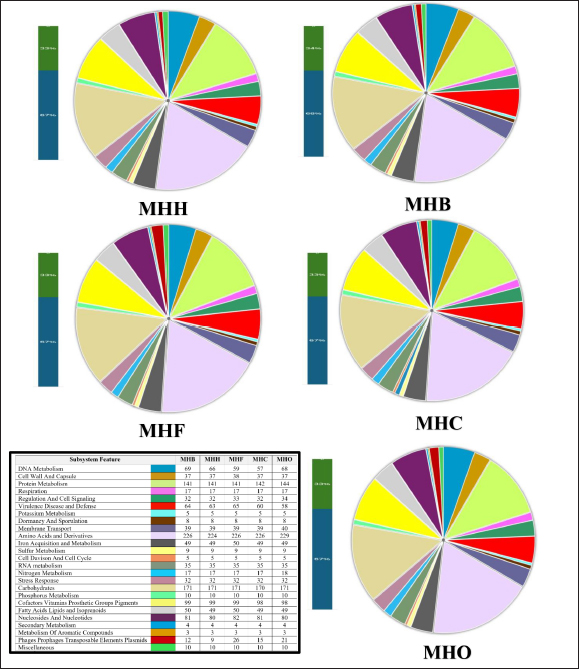
Fig. 3. An interactive genome comparison map between the human strain MHH (reference) and four animal strains was created using the RASTtk server’s Seed Viewer sequence-based comparison tool. The strains are (1) strain MHB, (2) strain MHC, (3) strain MHF, and (4) strain MHO, sorted from outer to inside rings. Colors range from 100% purple to 10% pale red, signifying the degree of amino acid similarity to the reference genome. The MHH reference strain’s genome is not shown in the graphic. Arrows (green, black, orange, purple, and blue) represent low-identity areas of CDs in the isolates compared to the MHH human strain. MLST has been used to categorize S. aureus isolates into clonal complexes (CCs) and STs. Certain pathogenic S. aureus complexes (CCs) and STs are particularly important as they affect humans and animals. The MLST server showed only seven registered Iraqi isolates, all of which are human isolates belonging to the clonal complexes CC1 (ST-1), CC5, and CC15 ( Jolley et al., 2018). These types are consistent with our MHH human strain, which is also a member of the ST-1group. However, the isolates from the animals were identified as MHC ST-1156, MHB ST-6, and MHF ST-6, while MHO, a new novel ST is closely related to ST 522. Many studies have reported these clonal complexes in humans and animals which may indicate zoonotic relationships. For example, researchers have reported the presence of ST-1156 in rodents (Ge et al., 2019), surgical wards (Aklilu et al., 2012), camel meat (Raji et al., 2016) , and humans (Abrudan et al., 2023; Roy et al., 2024). Whereas ST-6 were detected in humans (Roy et al., 2024) and in domestic animals and nonhuman primates (Schaumburg et al., 2015). However, the genome wide analyses provide better resolution compared to MLST as it provides detailed information to better understand strain-to-strain relationships (Rodrigues et al., 2022; Al-Tameemi et al., 2023). The RASTtk server sequence-based comparison tool and the TYGS demonstrated that while there were minor variations between the human strain MHH and the animals’ isolates, their genomes exhibited substantial similarity and formed a distinct cluster (Figs. 3 and 4). These findings may indicate a zoonotic relationship between human and animal isolates. The genomes of cow isolate MHB and cat isolate MHF showed significant homology to the human isolates of S. aureus (N315) (Kuroda et al., 2001) and the type strain of S. aureus (DSM 20231) (Shiroma et al., 2015). The genomes of the human isolate MHH and the dog isolate MHC were remarkably comparable to those of S. aureus (N315). Nevertheless, the MHO strain exhibited a lesser degree of similarity in comparison to the human and other isolates and was close to S. aureus subsp. anaerobius which was isolated originally from an abscess of a sheep with Morel’s disease in Khartoum state, Sudan (Elbir et al., 2013). It is worth mentioning that unlike the human strain MHH (which was used as the reference genome in the RASTtk analysis), the genomes of the animal isolates featured regions of clustered genes that had no matches or had minimal similarities. Figure 3 illustrates these regions, marked by black, green, orange, and blue arrows. These gene clusters may suggest the occurrence of horizontal gene transfer or gene deletion events. The MHB strain, in contrast to the other four analyzed isolates, exhibits a region (about 5727 base pairs-data not shown) with less similarity to the other isolates. The RASTtk server identifies this region as a cluster of proteins related to phages, with a similarity of 98–100% (Fig. 3, black arrow). The orange arrow indicates a region of putative proteins linked to S. aureus pathogenicity islands (SaPIs). SaPIs are small genetic elements that play a crucial role in the pathogenesis and virulence of the bacteria. They carry genes for superantigens, toxins, and other virulence factors, including the gene for toxic shock syndrome toxin. SaPIs are mobilized by specific bacteriophages (phages) through a process called lateral transduction, which allows them to transfer not only their own genes but also additional virulence genes from the host chromosome (Tormo et al., 2008; Novick and Ram, 2017). Except for the MHO strain, there were no matches or significant similarity found for the SaPI-related genes in the MHF, MHC, and MHB isolates (data not shown). SaPI-related genes are critical for S. aureus pathogenesis and pathogenicity. These genes encode superantigens, toxins, and other features that contribute to S. aureus ability to cause disease, including the gene responsible for producing toxic shock syndrome toxin (Tormo et al., 2008; Novick and Ram, 2017). The green arrow refers to a region with hypothetical proteins that have low identity to the MHH strain; however, the MHF and MHB strains exhibited genes that are identical to the mecA gene cassette (data not shown). The blue arrow denotes a section of hypothetical proteins with unknown roles in the MHH human strain that are not present in animal isolates. We are currently conducting an additional study to investigate the antimicrobial characteristics and the mobile genetic components associated with these regions.
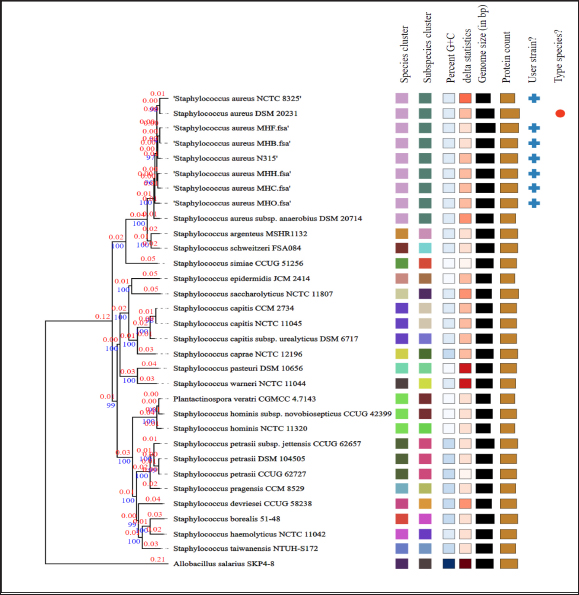
Fig. 4. Phylogenetic tree based on TYGS results for the local S. aureus whole-proteom. FastME 2.1.6.1 (Lefort et al., 2015) generated the tree using whole-proteome-based GBDP distances (genome BLAST distance phylogeny method). The lengths of the branches are scaled using the GBDP distance formula d5. The branch values (numbers above branches) are GBDP pseudo-bootstrap support values that are above 60% from 100 replications, with an average branch support of 92.0%. The tree was midpoint-rooted (Farris, 1972). Table 2. Biofilm associated genes in S. aureus MHH, MHB, MHF, MHC, and MHO isolates.
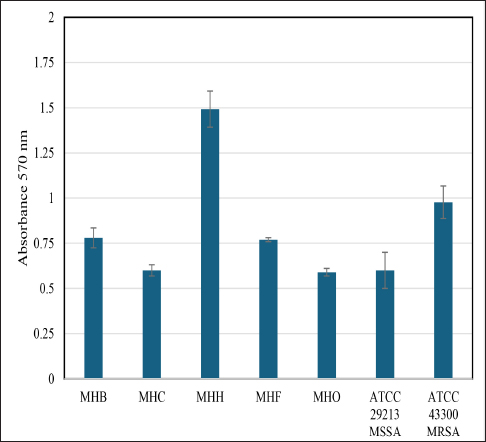
The process of biofilm formation in S. aureus is sophisticated and requires the coordinated expression of several genes. The ability to form biofilms differs among the various genetic lineages of S. aureus (Tasse et al., 2018). This is precisely what we noted, as illustrated in Figure 5. Our local isolates from diverse host sources displayed different levels of biofilm production, even though they shared the same biofilm-associated genes and regulators. The study by Croes et al., (2009) points out that some lineages, especially those assigned to MLST clonal complexes (like CC8), are more likely to produce more persistent biofilms than others. The reasons for variability among different S. aureus lineages are not fully understood but could be related to differences in gene expression, the presence of virulence factors, or environmental conditions(Tasse et al., 2018). Whatever the cause, these findings signify the capacity of certain lineages of S. aureus to cause disease (Croes et al., 2009; Idrees et al., 2021).
Fig. 5. Crystal violet binding assay showing biofilm formation ability of different S. aureus local isolates (MHB, MHC, MHH, MHF, and MHO) to the 96-well polystyrene plate surface in brain heart broth. Values represent the mean of two independent experiments; error bars indicate the standard deviation. S. aureus ATCC 29213, ATCC 43300 were used as a control. The main limitation of this study is the number of WGS samples included from each targeted host and the difficulty in proving the impact of genetic drift, particularly in biofilm-related genes, on the ability of bacteria to adapt to new animal host species through in vivo experiments. Previous investigations into a few in vivo models, including human-rabbit, human-ovine, and human-murine models, yielded varying conclusions, indicating the occurrence of multiple adaptation pathways in these models. The understanding of adaptive mechanisms that underlie species-specific pathogenesis could be improved further through the exploitation of host-switching models of pathogenesis (Viana et al., 2015; Bacigalupe et al., 2019; Mrochen et al., 2020). Consequently, the small number of samples from five distinct hosts in our analysis made it challenging to identify specific host adaptation genes across our targeted genomes. Consequently, we utilized the phylogenetic relationships and the overall genome identity to demonstrate the zoonotic nature of the isolates under investigation, as has been done in other studies (Abouelkhair and Kania, 2024; Jablonska et al., 2024; Wokorach et al., 2024). Due to the fact that biofilm formation in S. aureus is a crucial virulence trait that helps in colonization and adaptation inside the host during infection, we believe that tracing these genes in future zoonotic and host tropism studies is a valued study proposal that can be performed on local genomes. Unfortunately, there are no S. aureus whole genomic investigations in Iraq that we can compare or contrast in order to confirm this hypothesis. The magnitude and molecular basis of host-switching events are still inadequately understood, and a large-scale, genome-based analysis of the evolutionary history of S. aureus in the context of its host ecology is lacking. Our study is the first to include complete genome sequencing of S. aureus in Iraq, allowing us to annotate the biofilm-associated genes present in our local isolates. We conducted a thorough analysis of genes and regulators associated with biofilms in S. aureus, as described in the literature. In conclusion, this study mapped the genomes of local isolates for the first time, underscored the importance of periodic WGS for local isolates, identified genomic similarities among S. aureus local isolates indicating potential zoonotic relationships, and presented a list of biofilm-related genes for further exploration as critical virulence factors and potential determinants of host tropism. AcknowledgmentWe would like to thank Basrah University for providing the necessary resources to complete this work, as well as Biovet Laboratories at Basrah for providing the technical support to conduct this project. Conflict of interestThe Authors declare that there is no conflict of interest. FundingThis research did not receive specific funding. All expenses were covered by the authors. The initial work and genomic isolation were conducted at University of Basrah laboratories. Authors’ contributionsMohammed A. Al-Bukhalifa : Data curation; Formal analysis; Investigation, Methodology; review & editing. Hassan M. Al-Tameemi : Writing - original draft Data availabilityThe genome sequence of Staphylococcus aureus (MHB, MHH, MHO, MHF, MHC) has been deposited at GenBank under the accession numbers listed below.
The annotation was added by the NCBI Prokaryotic Genome Annotation Pipeline (PGAP-https://www.ncbi.nlm.nih.gov/genome/annotation_prok/). ReferencesAboud, W.A. and Khudaier, B.Y. 2018. Molecular detection of methicillin-resistant Staphylococcus aureus isolated from milk and cheese of cow and buffaloes in Basrah city. Basrah J. Vet. Res. 17, 806–819. Abouelkhair, M.A. and Kania, S.A. 2024. Whole genome sequencing and comparative genomics of six Staphylococcus schleiferi and Staphylococcus coagulans isolates. Genes 15(3), 284. Abrudan, M.I., Shamanna, V., Prasanna, A., Underwood, A., Argimón, S., Nagaraj, G. and Di Gregorio, S. 2023. Novel multidrug-resistant sublineages of Staphylococcus aureus clonal complex 22 discovered in India. MSphere 8(5), e0018523. Aklilu, E., Zakaria, Z., Hassan, L. and Cheng, C.H. 2012. Molecular relatedness of methicillin-resistant S. aureus isolates from staff, environment, and pets at University Veterinary Hospital in Malaysia. PLoS One 7(8), e43329. Al-Tameemi, H.M., Al-Hraishawi, H., Al-Hejjaj, M.Y., Abdulah, N.S., Alrafas, H.R. and Dawood, Y.A.H. 2023. Whole genome sequence and comparative genomics analysis of multidrug-resistant Staphylococcus xylosus NM36 isolated from a cow with mastitis in Basrah City. J. Genet. Eng. Biotechnol. 21(1), 163. Andrews, S. 2010. Babraham bioinformatics . FastQC: a quality control tool for high throughput sequence data. Available via https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ Archer, G.L. 1998. Staphylococcus aureus: a well-armed pathogen. Clin. Infect. Dis. 26(5), 1179–1181. Arciola, C.R., Campoccia, D., Gamberini, S., Baldassarri, L. and Montanaro, L. 2005. Prevalence of Cna, FnbA, and FnbB adhesin genes among Staphylococcus aureus isolates from orthopedic infections associated with different types of implant. FEMS Microbiol. Lett. 246(1), 81–86. Arciola, C.R., Campoccia, D., Speziale, P., Montanaro, L. and Costerton, J.W. 2012. Biofilm formation in Staphylococcus implant infections: a review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 33(26), 5967–5982. Aubourg, M., Pottier, M., Léon, A., Bernay, B., Dhalluin, A., Cacaci, M. and Torelli, R. 2022. Inactivation of the response regulator AgrA has a pleiotropic effect on biofilm formation, pathogenesis, and stress response in Staphylococcus lugdunensis. Microbiol. Spectr. 10(1), e0159821. Bacigalupe, R., Tormo-Mas, M.A., Penadés, J.R. and Fitzgerald, J.R. 2019. A multihost bacterial pathogen overcomes continuous population bottlenecks to adapt to new host species. Sci. Adv. 5(11), eaax0063. Bagger, F.O., Borgwardt, L., Jespersen, A.S., Hansen, A.R., Bertelsen, B., Kodama, M. and Nielsen, F.C. 2024. Whole genome sequencing in clinical practice. BMC Med. Genomics 17(1), 39. Bankevich, A., Nurk, S., Antipov, D., Gurevich, A.A., Dvorkin, M., Kulikov, A.S. and Lesin, V.M. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19(5), 455–477. Bartels, M.D., Worning, P., Andersen, L.P., Bes, M., Enger, H., Ås, C.G. and Hansen, T.A. 2021. Repeated introduction and spread of the MRSA clone T304/ST6 in Northern Europe. Clin. Microbiol. Infect. 27(2), 284.e1–284.e5. Becker, K. 2018. Pathogenesis of Staphylococcus aureus. In Staphylococcus aureus. Amsterdam, The Netherlands: Elsevier Inc. Beier, D. and Gross, R. 2006. Regulation of bacterial virulence by two-component systems. Curr. Opin. Microbiol. 9(2), 143–152. Bolger, A.M., Lohse, M. and Usadel, B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15), 2114–2120. Brettin, T., Davis, J., Disz, T., Edwards, R.A., Gerdes, S., Olsen, G.J., Olson, R., Overbeek, R., Parrello, B., Pusch, G.D., Shukla, M., Thomason, J.A. 3rd, Stevens, R., Vonstein, V., Wattam, A.R. and Xia, F. 2015. RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5, 8365; doi:10.1038/srep08365. Brlek, P., Bulić, L., Bračić, M., Projić, P., Škaro, V., Shah, N., Shah, P. and Primorac, D. 2024. Implementing whole genome sequencing (WGS) in clinical practice: advantages, challenges, and future perspectives. Cells 13(6), 504. Cheung, A.L., Nishina, K.A., Trotonda, M.P. and Tamber, S. 2008. The SarA protein family of Staphylococcus aureus. Int. J. Biochem. Cell Biol. 40(3), 355–361. Croes, S., Deurenberg, R.H., Boumans, M.L.L., Beisser, P.S., Neef, C. and Stobberingh, E.E. 2009. Staphylococcus aureus biofilm formation at the physiologic glucose concentration depends on the S. aureus lineage. BMC Microbiol. 9(1), 229. Cue, D., Junecko, J.M., Lei, M.G., Blevins, J.S., Smeltzer, M.S. and Lee, C.Y. 2015. SaeRS-dependent inhibition of biofilm formation in Staphylococcus aureus Newman. PLoS One 10(4), e0123027. Cue, D., Lei, M.G. and Lee, C.Y. 2012. Genetic regulation of the intercellular adhesion locus in staphylococci. Front. Cell Infect. Microbiol. 2, 38. Ebani, V.V. 2020. Biology and pathogenesis of Staphylococcus infection. Microorganisms 8(3), 11–13. Elbir, H., Robert, C., Nguyen, T.T., Gimenez, G., El Sanousi, S.M., Flock, J.I., Raoult, D. and Drancourt, M. 2013. Staphylococcus aureus subsp. anaerobius strain ST1464 genome sequence. Stand. Genomic Sci. 9(1), 1–13. Ewing, B. and Green, P. 1998. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8(3), 186–194. Farris, J.S. 1972. Estimating phylogenetic trees from distance matrices. Am. Nat. 106, 645–668. Frank, J.A., Reich, C.I., Sharma, S., Weisbaum, J.S., Wilson, B.A. and Olsen, G.J. 2008. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 74(8), 2461–2470. Frankel, M.B., Hendrickx, A.P.A., Missiakas, D.M. and Schneewind, O. 2011. LytN, a murein hydrolase in the cross-wall compartment of Staphylococcus aureus, is involved in proper bacterial growth and envelope assembly. J. Biol. Chem. 286(37), 32593–32605. Ge, J., Zhong, X., Xiong, Y., Qiu, M., Huo, S., Chen, X., Mo, Y., Cheng, M. and Chen, Q. 2019. Methicillin-resistant Staphylococcus aureus among urban rodents, house shrews, and patients in Guangzhou, Southern China. BMC Vet. Res. 15(1), 260; doi:10.1186/s12917-019-2012-8. Grande, R., Nistico, L., Sambanthamoorthy, K., Longwell, M., Iannitelli, A., Cellini, L., Di Stefano, A., Hall Stoodley, L. and Stoodley, P. 2014. Temporal expression of AgrB, CidA, and AlsS in the early development of Staphylococcus aureus UAMS-1 biofilm formation and the structural role of extracellular DNA and carbohydrates. Pathog. Dis. 70(3), 414–422. Gries, C.M., Biddle, T., Bose, J.L., Kielian, T. and Lo, D.D. 2020. Staphylococcus aureus fibronectin binding protein A mediates biofilm development and infection. Infect. Immun. 88(5), e00859–19. Hao, Z., Guo, Y., Rao, L., Yu, J., Zhan, Q., Xu, Y., Wang, B., Wu, X. and Yu, F. 2021. Deletion of Sarx decreases biofilm formation of Staphylococcus aureus in a polysaccharide intercellular adhesin (Pia)-dependent manner by downregulating Spa. Infect. Drug Resist. 14, 2241–2250. Hasani, A., Dehghani, L., Soltani, E. and Ebrahimzadeh Leylabadlo, H. 2023. Assessment of the presence of Sas family genes and their relationship with biofilm formation among clinical Staphylococcus aureus isolates. Pharm. Sci. 29(1), 46–51. Hong, J. and Roh, E. 2018. Complete genome sequence of biofilm-producing strain Staphylococcus xylosus S170. Korean J. Microbiol. 54(2), 167–68. Howden, B.P., Giulieri, S.G., Wong Fok Lung, T., Baines, S.L., Sharkey, L.K., Lee, J.Y.H., Hachani, A., Monk, I.R. and Stinear, T.P. 2023. Staphylococcus aureus host interactions and adaptation. Nat. Rev. Microbiol. 21(6), 380–395. Humphreys, H. and Coleman, D.C. 2019. Contribution of whole-genome sequencing to understanding of the epidemiology and control of meticillin-resistant Staphylococcus aureus. J. Hosp. Infect. 102(2), 189–199. Idrees, M., Sawant, S., Karodia, N. and Rahman, A. 2021. Staphylococcus aureus biofilm: morphology, genetics, pathogenesis and treatment strategies. Int. J. Environ. Res. Public Health 18(14), 7602. Jablonska, S., Kula, A. and Putonti, C. 2024. Draft genome of Staphylococcus capitis O112, isolated from the cheek swab of a healthy female. Microbiol. Resour. Announc. 13(3), e0127123. Jefferson, K.K., Pier, D.B., Goldmann, D.A. and Pier, G.B. 2004. The teicoplanin-associated locus regulator (TcaR) and the intercellular adhesin locus regulator (IcaR) are transcriptional inhibitors of the Ica locus in Staphylococcus aureus. J. Bacteriol. 186(8), 2449–2456. Jolley, K.A., Bray, J.E. and Maiden, M.C.J. 2018. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3, 124. Jolley, K.A. and Maiden, M.C.J. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11(1), 595. Kalantar-Neyestanaki, D., Mansouri, S., Tadjrobehkar, O. and Isaei, E. 2023. The frequency of adherence, biofilm-associated, arginine catabolic mobile element genes, and biofilm formation in clinical and healthcare worker coagulase-negative staphylococci isolates. BMC Microbiol. 23(1), 222. Keats, J.J., Cuyugan, L., Adkins, J. and Liang, W.S. 2018. Whole genome library construction for next generation sequencing. Methods Mol. Biol. 1706, 151–161. Kolar, S.L., Nagarajan, V., Oszmiana, A., Rivera, F.E., Miller, H.K., Davenport, J.E. and Riordan, J.T. 2011. NsaRS is a cell-envelope-stress-sensing two-component system of Staphylococcus aureus. Microbiology 157(Pt 8), 2206–2219. Kuehl, R., Al-Bataineh, S., Gordon, O., Luginbuehl, R., Otto, M., Textor, M. and Landmann, R. 2009. Furanone at subinhibitory concentrations enhances staphylococcal biofilm formation by LuxS repression. Antimicrob. Agents Chemother. 53(10), 4159–4166. Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I. and Cui, L. 2001. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357(9264), 1225–1240. Kwong, J.C., McCallum, N., Sintchenko, V. and Howden, B.P. 2015. Whole genome sequencing in clinical and public health microbiology. Pathology 47(3), 199–210. Larsen, M.V., Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R.L. and Jelsbak, L. 2012. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 50(4), 1355–1361. Lefort, V., Desper, R. and Gascuel, O. 2015. FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program: table 1. Mol. Biol. Evol. 32(10), 2798–2800. Lehman, M.K., Bose, J.L., Sharma-Kuinkel, B.K., Moormeier, D.E., Endres, J.L., Sadykov, M.R., Biswas, I. and Bayles, K.W. 2015. Identification of the amino acids essential for LytSR-mediated signal transduction in Staphylococcus aureus and their roles in biofilm-specific gene expression. Mol. Microbiol. 95(4), 723–737. Li, H. 2013. Aligning sequence reads, clone sequences, and assembly contigs with BWA-MEM. arXiv: Genomics; doi:10.6084/m9.figshare.963153.v1. Maljkovic Berry, I., Melendrez, M.C., Bishop-Lilly, K.A., Rutvisuttinunt, W., Pollett, S., Talundzic, E., Morton, L. and Jarman, R.G. 2019. Next generation sequencing and bioinformatics methodologies for infectious disease research and public health: approaches, applications, and considerations for development of laboratory capacity. J. Infect. Dis. 221(Suppl. 3), S292–S307. Marchler-Bauer, A., Derbyshire, M.K., Gonzales, N.R., Lu, S., Chitsaz, F., Geer, L.Y. and Geer, R.C. 2015. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 43(D1), 6. Mashruwala, A.A., Gries, C.M., Scherr, T.D., Kielian, T. and Boyd, J.M. 2017a. SaeRS is responsive to cellular respiratory status and regulates fermentative biofilm formation in Staphylococcus aureus. Infect. Immun. 85(8), e00157–17. Mashruwala, A.A., van de Guchte, A. and Boyd, J.M. 2017b. Impaired respiration elicits SrrAB-dependent programmed cell lysis and biofilm formation in Staphylococcus aureus. Elife 6, e23845. Mazaal, M., Ibrahim, H. and Mater, A. 2021. Molecular detection of nuc and sea genes of Staphylococcus aureus isolated from cow and sheep meat in Basrah city. Basrah J. Vet. Res. 20(1), 138–147. Meier-Kolthoff, J.P., Sardà Carbasse, J., Peinado-Olarte, R.L. and Göker, M. 2022. TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50(D1), D801–D807. Monk, I.R., Shaikh, N., Begg, S.L., Gajdiss, M., Sharkey, L.K., Lee, J.Y.H. and Pidot, S.J. 2019. Zinc-binding to the cytoplasmic PAS domain regulates the essential WalK histidine kinase of Staphylococcus aureus. Nat. Commun. 10(1), 3067. Mrochen, D.M., Fernandes de Oliveira, L.M., Raafat, D. and Holtfreter, S. 2020. Staphylococcus aureus host tropism and its implications for murine infection models. Int. J. Mol. Sci. 21(19), 7061. Narongpun, P., Chanchaithong, P., Yamagishi, J., Thapa, J., Nakajima, C. and Suzuki, Y. 2023. Whole-genome investigation of zoonotic transmission of livestock-associated methicillin-resistant Staphylococcus aureus clonal complex 398 isolated from pigs and humans in Thailand. Antibiotics 12(12), 1745. Novick, R.P. and Ram, G. 2017. Staphylococcal pathogenicity islands . overs and shakers in the genomic firmament. Curr. Opin. Microbiol. 38, 197–204. O’Brien, L., Kerrigan, S.W., Kaw, G., Hogan, M., Penadés, J., Litt, D., Fitzgerald, D.J., Foster, T.J. and Cox, D. 2002. Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol. Microbiol. 44(4), 1033–1044. O’Connor, A.M., McManus, B.A. and Coleman, D.C. 2018. First description of novel arginine catabolic mobile elements (ACMEs) types IV and V harboring a Kdp operon in Staphylococcus epidermidis characterized by whole genome sequencing. Infect. Genet. Evol. 61, 60–66. O’Gara, J.P. 2007. Ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol. Lett. 270(2), 179–88. Olson, R.D., Assaf, R., Brettin, T., Conrad, N., Cucinell, C., Davis, J.J. and Dempsey, D.M. 2023. Introducing the bacterial and viral bioinformatics resource center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 51(D1), 89. Overbeek, R., Olson, R., Pusch, G.D., Olsen, G.J., Davis, J.J., Disz, T., Edwards, R.A., Gerdes, S., Parrello, B., Shukla, M., Vonstein, V., Wattam, A.R., Xia, F. and Stevens, R. 2014. The SEED and the rapid annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42(D1), D206–D214; doi:10.1093/nar/gkt1226. Paharik, A.E. and Horswill, A.R. 2016. The staphylococcal biofilm: adhesins, regulation, and host response. Microbiol. Spectrum 4(2), 10.1128/microbiolspec.VMBF-0022-2015. Porayath, C., Suresh, M.K., Biswas, R., Nair, B.G., Mishra, N. and Pal, S. 2018. Autolysin mediated adherence of Staphylococcus aureus with fibronectin, gelatin and heparin. Int. J. Biol. Macromol. 110, 546–553. Prieto, J.M., Rapún-Araiz, B., Gil, C., Penadés, J.R., Lasa, I. and Latasa, C. 2020. Inhibiting the two-component system GraXRS with verteporfin to combat Staphylococcus aureus infections. Sci. Rep. 10(1), 17939. Raji, M.A., Garaween, G., Ehricht, R., Monecke, S., Shibl, A.M. and Senok, A. 2016. Genetic characterization of Staphylococcus aureus isolated from retail meat in Riyadh, Saudi Arabia. Front. Microbiol. 7, 911. Richardson, E.J., Bacigalupe, R., Harrison, E.M., Weinert, L.A., Lycett, S., Vrieling, M. and Robb, K., 2018. Gene exchange drives the ecological success of a multi-host bacterial pathogen. Nat Ecol. Evol. 2(9), 1468–1478. Rodrigues, R.A., Pizauro, L.J.L., Varani, A.M., Almeida, C.C., Silva, S.R., Cardozo, M.V., MacInnes, J.I., Kropinski, A.M., Melo, P.C. and Ávila, F.A. 2022. Comparative genomics study of Staphylococcus aureus isolated from cattle and humans reveals virulence patterns exclusively associated with bovine clinical mastitis strains. Front. Microbiol. 13, 1033675. Roy, S., Aung, M.S., Paul, S.K., Nasreen, S.A., Haque, N., Mazid, R. and Khan, M.S. 2024. Genetic characterization of methicillin-resistant/susceptible Staphylococcus aureus (MRSA/MSSA) and Staphylococcus argenteus clinical isolates in Bangladesh: dominance of ST6-MRSA-IV/T304 and detection of Cfr/FexA in ST8-MSSA/T008. IJID Regions. 10, 132–139. Schaumburg, F., Pauly, M., Anoh, E., Mossoun, A., Wiersma, L., Schubert, G. and Flammen, A. 2015. Staphylococcus aureus complex from animals and humans in three remote African regions. Clin. Microbiol. Infect. 21(4), 345.e1–8. Schroeder, K., Jularic, M., Horsburgh, S.M., Hirschhausen, N., Neumann, C., Bertling, A. and Schulte, A. 2009. Molecular characterization of a novel Staphylococcus aureus surface protein (SasC) involved in cell aggregation and biofilm accumulation. PLoS One 4(10), e7567. Shareef, A.A., Farhan, F.J. and Alriyahee, F.A.A. 2023. Antibacterial activity of silver nanoparticles composed by fruit aqueous extract of Abelmoschus esculentus (L.) Moench alone or in combination with antibiotics. Basrah J. Agric. Sci. 36(2), 144–174. Sharma-Kuinkel, B.K., Mann, E.E., Ahn, J.S., Kuechenmeister, L.J., Dunman, P.M. and Bayles, K.W. 2009. The Staphylococcus aureus LytSR two-component regulatory system affects biofilm formation. J. Bacteriol. 191(15), 4767–4775. Shaw, L.N., Golonka, E., Szmyd, G., Foster, S.J., Travis, J. and Potempa, J. 2005. Cytoplasmic control of premature activation of a secreted protease zymogen: deletion of Staphostatin B (SspC) in Staphylococcus aureus 8325-4 yields a profound pleiotropic phenotype. J. Bacteriol. 187(5), 1751–1762. Shiroma, A., Terabayashi, Y., Nakano, K., Shimoji, M., Tamotsu, H., Ashimine, N. and Ohki, S. 2015. First complete genome sequences of Staphylococcus aureus subsp. aureus Rosenbach 1884 (DSM 20231 T), determined by PacBio single-molecule real-time technology. Genome Announc. 3(4), e00800–15. Srinivasan, R., Karaoz, U., Volegova, M., MacKichan, J., Kato-Maeda, M., Miller, S., Nadarajan, R., Brodie, E.L. and Lynch, S.V. 2015. Use of 16S rRNA gene for identification of a broad range of clinically relevant bacterial pathogens. PLoS One 10(2), 1–22. Talat, A., Khalid, S., Majeed, H.A. and Khan, A.U. 2020. Whole-genome sequence analysis of multidrug-resistant Staphylococcus epidermidis ST35 strain isolated from human ear infection of an Iraqi patient. J. Glob. Antimicrob. Resist. 21, 318–320. Tan, L., Huang, Y., Shang, W., Yang, Y., Peng, H., Hu, Z. and Wang, Y. 2022. Accessory gene regulator (Agr) allelic variants in cognate Staphylococcus aureus strain display similar phenotypes. Front. Microbiol. 13, 700894. Tasse, J., Trouillet-Assant, S., Josse, J., Martins-Simões, P., Valour, F., Langlois-Jacques, C. and Badel-Berchoux, S. 2018. Association between biofilm formation phenotype and clonal lineage in Staphylococcus aureus strains from bone and joint infections. PLoS One 13(8), e0200064. Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E.P., Zaslavsky, L., Lomsadze, A., Pruitt, K.D., Borodovsky, M. and Ostell, J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44(14), 6614–6624. Thamer M.A and Shareef A.A. 2022. Molecular detection of some vancomycin pathogenic bacteria in Basrah governorate. Basrah Res. Sci. 12, 27–34. Thiran, E., Di Ciccio, P.A., Graber, H.U., Zanardi, E., Ianieri, A. and Hummerjohann, J. 2018. Biofilm formation of Staphylococcus aureus dairy isolates representing different genotypes. J. Dairy Sci. 101(2), 1000–1012. Tiwari, N., Lopez-Redondo, M.L., Miguel-Romero, L., Kulhánková, K., Cahill, M.P., Tran, P.M. and Kinney, K.J. 2020. The SrrAB two-component system regulates Staphylococcus aureus pathogenicity through redox-sensitive cysteines. Proc. Natl. Acad. Sci. U. S. A. 117(20), 11256–11264. Tormo, M.A., Ferrer, M.D., Maiques, E., Úbeda, C., Selva, L., Lasa, I., Calvete, J.J., Novick, R.P. and Penadés, J.R. 2008. Staphylococcus aureus pathogenicity island DNA is packaged in particles composed of phage proteins. J. Bacteriol. 190(7), 2434–2440. Trotonda, M.P., Tamber, S., Memmi, G. and Cheung, A.L. 2008. MgrA represses biofilm formation in Staphylococcus aureus. Infect. Immun. 76(12), 5677–5686. Viana, D., Comos, M., McAdam, P.R., Ward, M.J., Selva, L., Guinane, C.M., González-Muñoz, B.M., Tristan, A., Foster, S.J., Fitzgerald, J.R. and Penadés, J.R. 2015. A single natural nucleotide mutation alters bacterial pathogen host tropism. Nat. Genet. 47(4), 361–366. Vuppada, R.K., Hansen, C.R., Strickland, K.A.P., Kelly, K.M. and McCleary, W.R. 2018. Phosphate signaling through alternate conformations of the PstSCAB phosphate transporter. BMC Microbiol. 18(1), 37. Walker, J.N., Crosby, H.A., Spaulding, A.R., Salgado-Pabón, W., Malone, C.L., Rosenthal, C.B., Schlievert, P.M., Boyd, J.M. and Horswill, A.R. 2013. The Staphylococcus aureus ArlRS two-component system is a novel regulator of agglutination and pathogenesis. PLoS Pathog. 9(12), e1003819. Wattam, A.R., Davis, J.J., Assaf, R., Boisvert, S., Brettin, T., Bun, C., Conrad, N., Dietrich, E.M., Disz, T., Gabbard, J.L., Gerdes, S., Henry, C.S., Kenyon, R.W., Machi, D., Mao, C., Nordberg, E.K., Olsen, G.J., Murphy-Olson, D.E., Olson, R., Overbeek, R., Parrello, B., Pusch, G.D., Shukla, M., Vonstein, V., Warren, A., Xia, F., Yoo, H. and Stevens, R.L. 2017. Improvements to PATRIC, the all-bacterial bioinformatics database and analysis resource center. Nucleic Acids Res. 45(D1), D535–D542 Wokorach, G., Erima, B., Najjuka, F., Kiyengo, J., Kibuuka, H., Musinguzi, A.K., Wabwire-Mangen, F. and Byarugaba, D.K. 2024. Draft genome sequence of Staphylococcus urealyticus strain MUWRP0921, isolated from the urine of an adult female Ugandan. Microbiol. Resour. Announc. 13(1), e0081723; doi:10.1128/mra.00817-23. Yang, M., Derbyshire, M.K., Yamashita, R.A. and Marchler-Bauer, A. 2020. NCBI’s conserved domain database and tools for protein domain analysis. Curr. Protoc Bioinfor. 69(1), e90; doi:10.1002/cpbi.90; doi:10.1093/nar/gkw1017. | ||
| How to Cite this Article |
| Pubmed Style Al-bukhalifa MA, Al-tameemi HM. First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes. Open Vet. J.. 2024; 14(12): 3269-3288. doi:10.5455/OVJ.2024.v14.i12.12 Web Style Al-bukhalifa MA, Al-tameemi HM. First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes. https://www.openveterinaryjournal.com/?mno=216021 [Access: June 22, 2026]. doi:10.5455/OVJ.2024.v14.i12.12 AMA (American Medical Association) Style Al-bukhalifa MA, Al-tameemi HM. First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes. Open Vet. J.. 2024; 14(12): 3269-3288. doi:10.5455/OVJ.2024.v14.i12.12 Vancouver/ICMJE Style Al-bukhalifa MA, Al-tameemi HM. First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes. Open Vet. J.. (2024), [cited June 22, 2026]; 14(12): 3269-3288. doi:10.5455/OVJ.2024.v14.i12.12 Harvard Style Al-bukhalifa, M. A. & Al-tameemi, . H. M. (2024) First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes. Open Vet. J., 14 (12), 3269-3288. doi:10.5455/OVJ.2024.v14.i12.12 Turabian Style Al-bukhalifa, Mohammed A., and Hassan M. Al-tameemi. 2024. First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes. Open Veterinary Journal, 14 (12), 3269-3288. doi:10.5455/OVJ.2024.v14.i12.12 Chicago Style Al-bukhalifa, Mohammed A., and Hassan M. Al-tameemi. "First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes." Open Veterinary Journal 14 (2024), 3269-3288. doi:10.5455/OVJ.2024.v14.i12.12 MLA (The Modern Language Association) Style Al-bukhalifa, Mohammed A., and Hassan M. Al-tameemi. "First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes." Open Veterinary Journal 14.12 (2024), 3269-3288. Print. doi:10.5455/OVJ.2024.v14.i12.12 APA (American Psychological Association) Style Al-bukhalifa, M. A. & Al-tameemi, . H. M. (2024) First whole genome sequencing of Staphylococcus aureus isolates from Iraq: Insights into zoonotic relations and biofilm-related genes. Open Veterinary Journal, 14 (12), 3269-3288. doi:10.5455/OVJ.2024.v14.i12.12 |

